1. Построение дерева по нуклеотидным последовательностям
В данном задании необходимо построить филогенетическое дерево тех же бактерий, что и в предыдущих заданиях, использую последовательность РНК малой субъединицы рибосомы (16S rRNA).| Название | Мнемоника | AC записи EMBL | Координаты РНК | Цепь |
| Bacillus anthracis | BACAN | AE017334 | 29129..30635 | Прямая |
| Enterococcus faecalis | ENTFA | AE017334 | 248466..249987 | Прямая |
| Geobacillus kaustophilus | GEOKA | BA000043 | 10421..11973 | Прямая |
| Lactobacillus delbrueckii | LACDA | CR954253 | 45160..46720 | Прямая |
| Lactococcus lactis | LACLM | AM406671 | 45160..46720 | Прямая |
| Listeria monocytogenes | LISMO | AM406671 | 99187..100732 | Обратная |
| Staphylococcus epidermidis | STAES | AE015929 | 1598006..1599559 | Обратная |
Далее надо вырезать нужные участки из записи EMBL:
seqret -saskДалее, отвечая на вопросы seqret, надо ввести названеи файла ****.embl, начало и конец, а так же комплиментарна ли цепь.
Я поместила последовательность в один файл и отредактировала его, оставив только мнемонику видов. Затем необходимо провести выравнивание отобранных последовательностей белков.
muscle -in all.fasta -out align_all.fastaПолучила выравнивание в fasta формате.
Затем я открыла его в программе MEGA и построила дерево методом Neighbor joining:
| Полученное дерево | Исходное дерево |
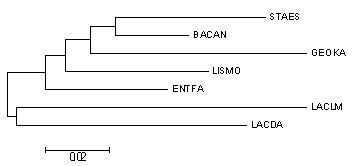
|
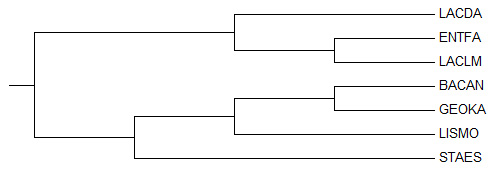
|
{LACLM; LACDA}vs{STAES; BACAN; GEOKA; LISMO; ENTFA};
{STAES; BACAN}vs{GEOKA; LISMO; ENTFA; LACLM; LACDA};
{STAES; BACAN; GEOKA}vs{LISMO; ENTFA; LACLM; LACDA};
2. Построение и анализ дерева, содержащего паралоги
Необходимо найти в выбранных бактериях достоверные гомологи белка CLPX_BACSU и построить дерево этих гомологов.seqret sw:clpx_bacsuДалее, воспользовавшись файлом proteo.fasta, я создала базу данных для поиска гомологов:
formatdb -i proteo.fasta -p T -n baseА затем провела поисз гомологов:
blastall -p blastp -d base -i clpx_bacsu.fasta -e 0.001 -o blastp.outЗатем из полученного и отредактированного файла выбрала нужные белки
Далее я создала лист-файл со списком идентификаторов.
Необходимо получить последовательности в fasta формате, поэтому я выполнила следущую команду:
seqret @my_hom.list my_hom.fastaа затем выровняла полученные последовательности
muscle -in my_hom.fasta -out align_my_hom.fastaПолученное выравнивание я открыла в программе MEGA и реконструировала методом Neighbor joining.
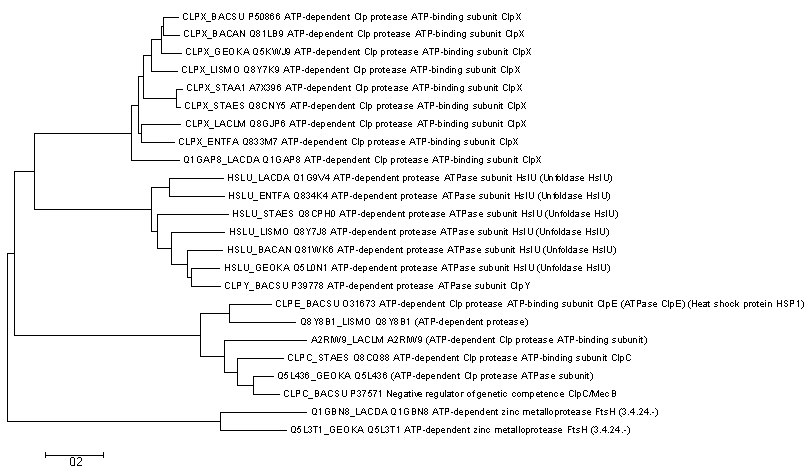
Два гомологичных белка будем называть ортологами, если они 1) из разных организмов и 2) разделение их общего предка на линии, ведущей к ним, произошла в результате видообразования
Два гомологичных белка из одного организма будем называть паралогами
Некоторые ортологи:
CLPX_BACSU, CLPX_BACAN
Q1GBN8_LACDA, Q5L3T1_GEOKAНекоторые парологи:
HSLU_GEOKA, CLPX_GEOKA
CLPX_LISMO, HSLU_LISMO