Молекулярная динамика биологических молекул в GROMACS
Целью этой работы является знакомство с возможностями моделирования молекулярной динамики.
В работе используются следующие типы файлов:
Моделирование перехода ДНК из А в В форму
Для работы даны следующие файлы:
В файле dna.pdb удаляем 5'-фосфаты, прибавляем к названиям нуклеотидов D и заменяем названия атомов C5M на C7. Cтроим файл топологии системы в силовом поле amber99sb и файл с координатами в формате Gromacs.
pdb2gmx -f dna.pdb -o dna -p dna -ff amber99sb -water tip3p
Делаем небольшой отступ в ячейке от ДНК
editconf -f dna.gro -o dna_ec -d 1.5
И проводим оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле
grompp -f em -c dna_ec -p dna -o dna_em -maxwarn 1 mdrun -deffnm dna_em -v
Начальное значение максимальной силы 4.810e+03 on atom 12
Конечное значение макимальной силы 1.323e+03 on atom 304
Добавляем в ячейку молекулы воды
genbox -cp dna_em -p dna -cs -o dna_s
И нейтрализуем заряд системы. Это делаем в два шага: строим tpr и запускаем genion
grompp -f em -p dna -c dna_s -o dna_s genion -s dna_s -o dna_si -p dna -np X
X - это количество положительных ионов необходимых для нейтрализации заряда системы, равно 10. Проводим "утряску" воды:
grompp -f pr -c dna_si -p dna -o dna_pr -maxwarn 1 mdrun -deffnm dna_pr -v
Переформатируем dna_pr.gro и dna_si.gro в pdb формат
editconf -f dna_pr.gro -o dna_pr.pdb editconf -f dna_si.gro -o dna_si.pdb
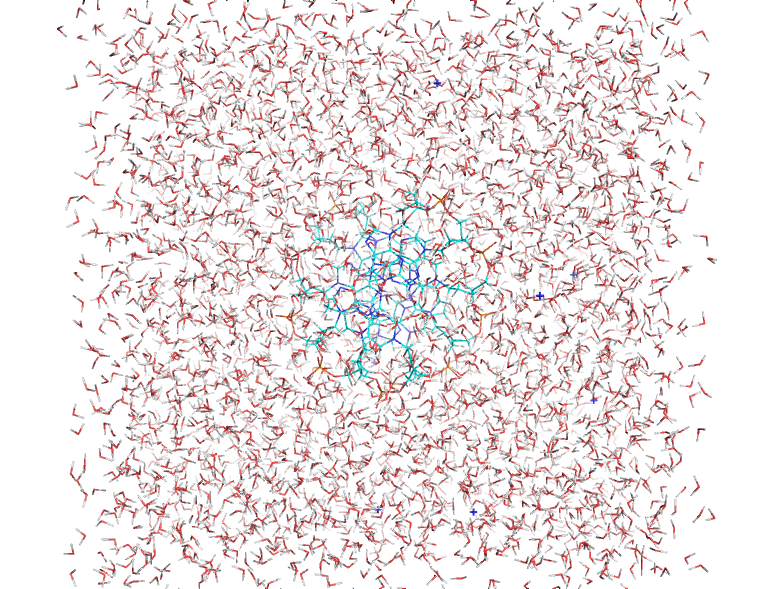
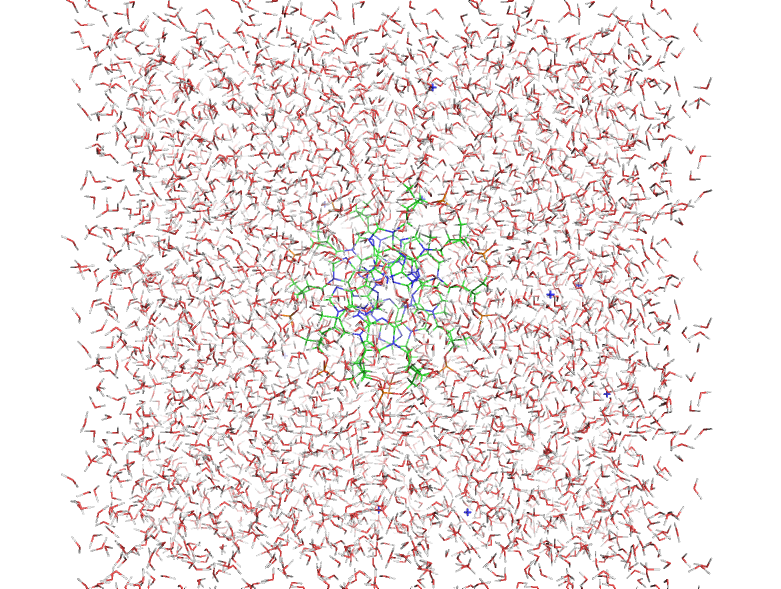
Слева изображение до "утряски", справа после. Как видно, в итоге вода расположена более упорядочно.
Суперкомпьютер
Первым делом копируем файлы
scp -r ./Atrokhova/ skif:_scratch/fbb
и запускаем тестовое моделирование на суперкомпьтере
ssh skif cd _scratch/fbb/Atrokhova cp /home/users/golovin/progs/share/gromacs/top/residuetypes.dat . cp -r /home/users/golovin/progs/share/gromacs/top/amber99sb.ff/ . grompp -f md -c dna_pr -p dna -o dna_md -maxwarn 1 sbatch -n 4 -e error.log -o output.log -t 5 -p test impi /mnt/msu/users/fbbstudent/gmx-4.6.7-intel-impi-mkl-single/bin/mdrun_mpi -deffnm dna_md -v
Получила номер, следить за ходом счета можно в файле error.log. Но так как задача тестовая, счет останавливается. Все хорошо.
Теперь запускаем основное моделирование
sbatch -N1 --ntasks-per-node=2 -e error-gpu.log -o output.log -t 350 -p gpu impi /opt/ccoe/gromacs-5.0.4/build/bin/gmx_mpi mdrun -testverlet -deffnm dna_md -v
Номер моей задачи: 1044993