Все выполненные команды в этом занятии:
pdb2gmx -f camelid.pdb -p -ignh pdb2gmx -f amylase.pdb -p -ignh # добавление водородов в структуры лиганда и рецептора
editconf -f conf.gro -o camelid_h.pdb editconf -f conf.gro -o amylase_h.pdb # конвертация файла выхода conf.gro в pdb
mark_sur amylase_h.pdb camelid_m.pdb mark_sur amylase_h.pdb amylase_m.pdb #добавление зарядов и радиусов к атомам
zdock -R amylase_m.pdb -L camelid_m.pdb # непосредственно докинг (работает около 12 мин), файл выхода zdock.out
1) в pdb файлы amylase_m.pdb camelid_m.pdb добавлены названия цепей А, В соотетственно. Они, видимо, пропали в conf.gro при добавлении Н.
2) в amylase_m.pdb camelid_m.pdb удалены все шапки, а непосредственно перед списком атомов есть 1 пустая строка
3) в amylase_m.pdb camelid_m.pdb оставлена последняя строка с ter, но по-моему их наличие вовсе не сыграло никакой роли в "запускаемости"
4) в zdock.out удалена 2 строка, в первой строке осталено только 2 первые значения: 128 1.2, а 0 удален
zrank zdock.out 1 2000 # считаем zrank-score для всех комплексов (с 1 по 2000), очень привередливая программа, файл выхода zdock.out.zr.out
cp /home/shad/progs/bin/create_lig .
create.pl zdock.out # создание pdb файлов комплексов амилазы и участка антитела ls complex*pdb>list zrank list # другой способ посчитать zrank-score
Затем сколько я ни пыталась запускать g_rms для сравнения, всегда получался один и тот же результат.
Опытным путем было установлено, что надо бы удалить из полученных комплексов водороды.
С помощью babel была поытка это совершить, но оказалось, что нужно сначала из всех комплексов нужно удалить первую пустую строку.
В с помощью script_1 все удалось: получился файл с номером комплекса и его rmsd.
Все полученные файлы
После добавления водородов в обе структуры: amylase_h.pdb , camelid_h.pdb
После проставления зарядов и радиусов атомов: amylase_m_1.pdb , camelid_m_1.pdb
После проставления зарядов и радиусов атомов (+ добавлены названия цепей): amylase_m.pdb , camelid_m.pdb
Файл-выхода докинга: zdock.out
Скрипт добавления к атомам названия цепи: script.py
Файл с zrank-score: zdock.out.zr.out
Пример файла комплекса до обработки: complex.1.pdb
Тот же файл комплекса после удаления водородов: 1_noh.pdb
Скрипт для получения rmsd: script_1
Файл с rmsd для всех комплексов: RMSD
Файл Exel для комплексов: zdock.xls
Результаты:
Zrank score дает оценку полученным комплексам:
Score=w(vdW_a)*E(vdW_a)+w(vdW_r)*E(vdW_r)+w(elec_sra)*E(elec_sra)+w(elec_srr)*E(elec_srr)+w(elec_lra)*E(elec_lra)+w(elec_lrr)*E(elec_lrr)+w(ds)*E(ds)
Оказалось, что 3 лучшими комплексами с точки зрения энергии являются: № 974, 938, 1841. На рисунке показаны фиолетовым - структура 1kxt, голубым -построенная в результате докинга
| Complex_974 | Complex_938 | Complex_1841 |
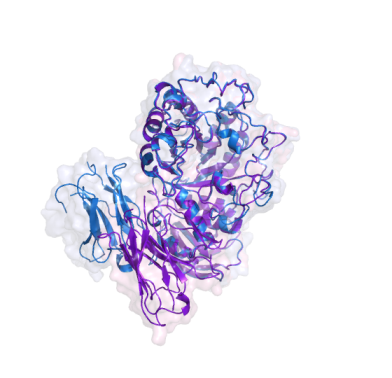
|
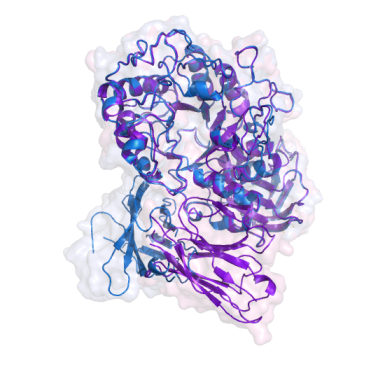
|
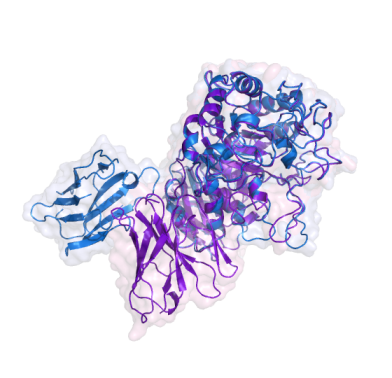
|
| Zrank-score -30.3291 |
Zrank-score -27.1881 |
Zrank-score -22.432 |
Всо всех полученных комплексах места связывания на поверхности амилазы располагаются близко к 1kxt, но ни одна структура не совпала с комплексом из pdb-банка.
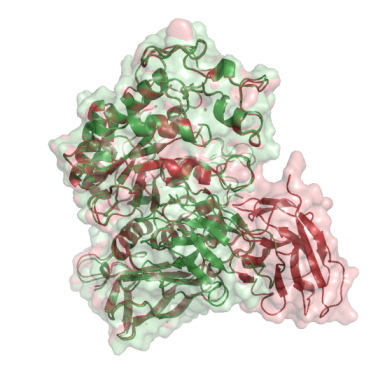
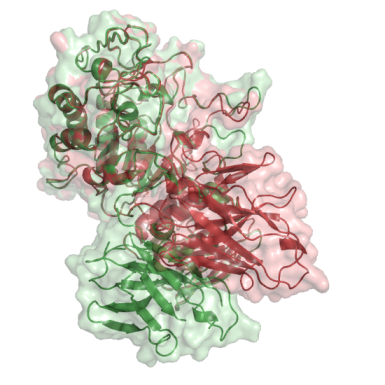
При сравнении с помощью g_rms, оказалось, что наименьшее отклонение от известной структуры имеют комплексы: № 1571, 1479, 1504. На рисунке показаны красным - структура 1kxt, зеленым - построенная в результате докинга
| Complex_1571 | Complex_1479 | Complex_1504 |
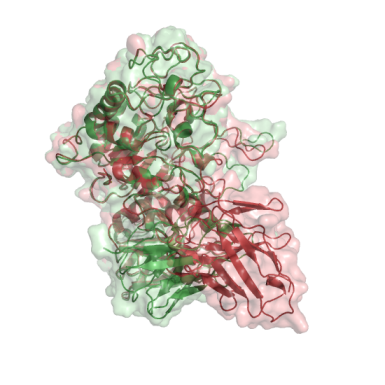
|
|
|
| RMSD 1,041683 |
RMSD 1,04565 |
RMSD 1,070853 |
В этих комплексах места связывания тоже примерно в том же самом месте, что и у 1kxt, но комплексы не выравниваются в участке антитела. Таким образом, неплохо бы было запустить докинг с параметром -d.
Оба сравниваемых параметов для всех изображенных комплексов:
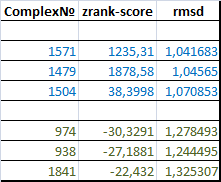
Таким образом, неплохо бы было запустить докинг с параметром -d.