Докинг низкомолекулярных лигандов в структуру белка
Для всех операций по моделированию комплекса лизоцима LYS_ANTMY с лигандом NAG воспользуемся структурой данного белка, предсказанной по гомологии, в которой удалены атомы лиганда: seq5.pdb
- Докинг низкомолекулярного лиганда NAG в структуру лизоцима
1. CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O - smiles нотация лиганда NAG.
При помощи команд:
obgen nag.smi > nag.mol
babel -imol nag.mol -opdb nag.pdb
babel -ipdb nag.pdb -omop nag.mop -xk "PM6"
MOPAC2009.exe nag.mop
babel -imopout nag.out -opdb nag.pdb
построили 3D модель лиганда NAG (nag.pdb) из его smiles нотации nag.smi
2. Скриптом prepare_ligand4.py из пакета Autodock tools создадим pdbqt файл лиганда (команда /home/preps/golovin/progs/bin/prepare_ligand4.py -l nag.pdb -o nag.pdbqt)
На выходе получаем pdbqt файл для лиганда.
3. Скриптом prepare_receptor4.py подготовим молекулу рецептора (команда /home/preps/golovin/progs/bin/prepare_receptor4.py -r seq5.pdb -o seq5.pdbqt)
На выходе получаем seq5.pdbqt файл.
4. Создаем файл с параметрами для докинга vina.cfg.
5. Проведем первый докинг с фиксированными аминокислотами белка и подвижным лигандом. Запустим команду vina --config vina.cfg --receptor seq5.pdbqt --ligand nag.pdbqt --out nag_prot.pdbqt --log nag_prot.log
На выходе имеем два файла nag_prot.pdbqt, содержащий информацию о положениях лиганда, и nag_prot.log, содержащий информацию о энергиях каждой модели.
Ниже на рисунке ппредставлены все состояния лиганда.
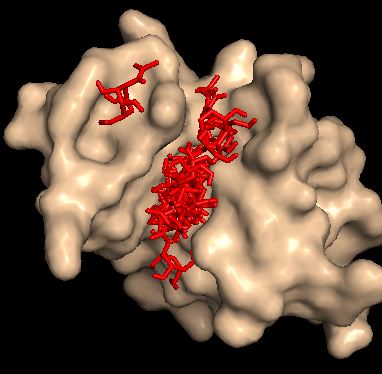 | 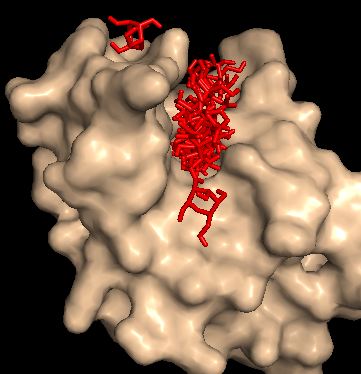 |
Видно, что лиганд может занимать любую позицию которую мы ему укажем (одна модель отражает положение наг'а в нестрандартном окружение (не природном)).
Ниже приведены модели с наилучшими энегиями (последняя (№15) - выбивающаяся модель).
| # модели | Аффинность (kcal/mol) | Геометрическая разница (rmsd u.b.) |
| 1 | -6,3 | 0.000 |
| 2 | -5.7 | 3.063 |
| 3 | -5.3 | 2.908 |
| 15 | -4.1 | 13.089 |
(видно, что положение вдали от нативного центра связывания энергетически менее выгодно).
6. Проведем докинг лиганда (подвижен) в белок, у которого подвижны некоторые боковые радикалы.
Задаем подвижную и константную часть при помощи команды python /usr/share/pyshared/AutoDockTools/Utilities24/prepare_flexreceptor4.py -r prot.pdbqt -s GLU32_ASN44_ASP50_ASN57_ALA102 (здесь мы указываем аминокислот, боковые цепи которых вращаются)
Затем запустим моделирование. в результате моделирования мы получили файлы nag1.pdbqt, nag1.log, аналогичные файлам в предыдущих пунктах Все состояния лиганда (красный) и выбранных аминокислотных остатков (зеленый) можно посмотреть ниже:
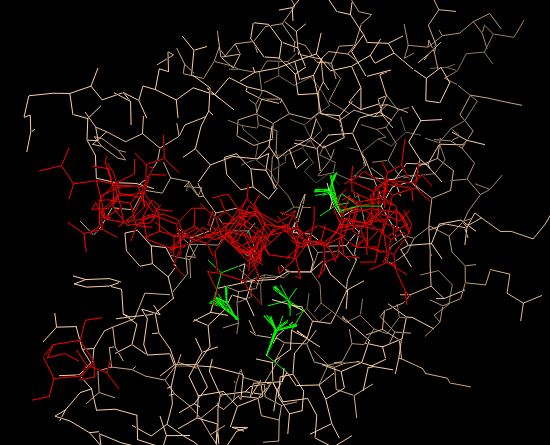
Далее в таблице представлены характеристики лучших моделей и сравнение этих параметров для докинга с неподвижным белком.
| # модели | Аффинность (подвижный белок) (kcal/mol) | Аффинность (неподвижный белок)(kcal/mol) | Геометрическая разница (rmsd u.b.) |
| 1 | -6,4 | -6,3 | 0.000 |
| 2 | -6,3 | -5.7 | 2.346 |
| 3 | -5,1 | -5.3 | 2.971 |
Из этих данных трудно что-либо сказать. где-то аффинность выросла, где-то упала, а где-то (модель 1) практические не изменилась. Но со стопроцентной вероятностью можно сказать, что аминокислотные радикалы крутятся).
Но, стоит отметить, несмотря на отсутствие явных преимуществ, существует плюс гибкого моделирования, заключающийся в следующем: в природе белки не являются неподвижными молекулами, при взаимодействии с лигандом может происходить не только вращение боковых радикалов (что мы и делаем при подобном типе докинга), но и изменение вторичной структуры белка. Так что, более правильно моделировать именно с подвижными боковыми радикалами (из соображений сходства подобной системы с белками в растворе).
7. Лиганд располагается наиболее близко к положению лиганда, полученного при моделировании белка по гомлогии, в моделе №20 (нпри моделировании с неподвижным белком) с энергией взаимодействия -3.7kcal/mol, а при моделировании с подвижными радикалами, не удалось обнаружить подобного варианта (есть много вариантов, когда лиганд лежит там же где и лиганд, полученный на основе гомологичного моделирования, но он сильно повернут). Но, стоит отметичть, что энергетическая невыгодность такого положения лиганда не указывает на низкую вероятность реализации подобного расположени, т.к. мы взяли в рассмотрение лишь один мономер из трисахарида. если посмотреть на предсказанную структуру (по гомологии), большая часть водородных связей (которые обеспечивают удержание лиганда)образована с атомами трисахарида, принадлежащими другуому моносахариду.неудивительно, что мы не видим правильного расположения лиганда (согласующегося с моделью по гомологии) при докинге. стоило бы в данной ситуации дочить трисахарид (или ту часть трисахарида, с которой образуется наибольшее количество водородных связей).
8. Произведем замену метильного радикала в боковой группе СH3C(=O)NH NAG'a на OH (nag_oh.smi), H (nag_h.smi), Ph (nag_ph.smi), NH2 (nag_nh2.smi). Запустим скрипт следующего содержания:
export PATH=${PATH}:/home/preps/golovin/progs/bin
for i in nag_h nag_oh nag_nh2 nag_ph ;do
obgen ${i}.smi > ${i}.mol
babel -imol ${i}.mol -opdb ${i}.pdb
babel -ipdb ${i}.pdb -omop ${i}.mop -xk "PM6"
MOPAC2009.exe ${i}.mop
babel -imopout ${i}.out -opdb ${i}1.pdb
/home/preps/golovin/progs/bin/prepare_ligand4.py -l ${i}1.pdb -o ${i}.pdbqt
vina --config vina.cfg --receptor seq5.pdbqt --ligand ${i}.pdbqt --out ${i}_seq5.pdbqt --log ${i}_seq5.log
vina --config vina.cfg --receptor seq5_rigid.pdbqt --flex seq5_flex.pdbqt --ligand ${i}.pdbqt --out ${i}_seq5_fr.pdbqt --log ${i}_seq5_fr.log
done
Все полученные файлы можн посмотреть по ссылке Practice9.
Проведем анализ результатов замен метильного радикала на прочие.
| №модели | CH3 | CH3_flex | H | H_flex | OH | OH_flex | NH2 | NH2_flex | Ph | Ph_flex |
| 1 | -6,3 | -6,4 | -5,5 | -5,4 | -1,3 | -1,2 | -6,3 | -6,1 | -7 | -7,2 |
| 2 | -5,7 | -6,3 | -5,1 | -5,2 | -1,1 | -1,2 | -5,8 | -5,9 | -6,3 | -6,1 |
| 3 | -5,3 | -5,1 | -4,8 | -4,5 | -1,1 | -1,2 | -5,5 | -5 | -6,2 | -6 |
Ниже на графике приведено сравнение энергий моделей с номерами 1-20 ({i}_FLEX - результаты моделирования с подвижными аминокислотными остатками)
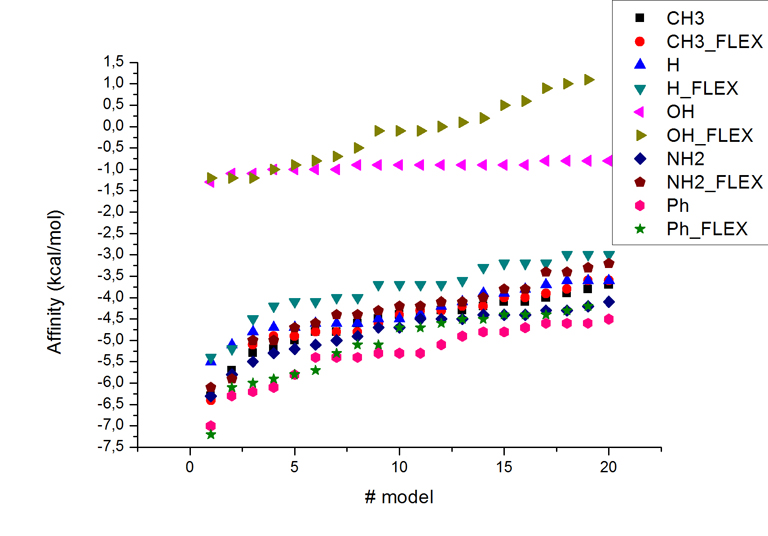
Из данных таблицы и графика можно сделать следующие выводы:
- Значени энергий связывания лиганда при моделировании с подвижными боковыми радикалами (выбранными нами заранее) практически всегда хуже, чем в случае моделирования с неподвижным белком.
- Замена метильного радикала на гидроксил приводит к "потери сродства" лиганда к данному центру связывания.
- Замена метильного радикала на водород снижает сродство лиганда к сайту связывания.
- Замена метильного радикала на аминогруппу практически не меняет сродство лиганда к сайту связывания.
- Замена метильного радикала на фенил существенно повышает сродство лиганда к сайту связывания.