Abinitio вычисления для нафталина и азулена
- Оптимизировать с помощью MOPAC структуры нафталена и азулена ниже приведены результаты оптимизации структур азулена(azulene.smi) и нафталина (napthalene.smi) при помощи obgen:
- Оптимизация с помощью GAMESS
- Расчёт энергии методом Хатри-Фока и методом теории функционала плотности
- Сравнение энергий, полученных методом Хатри-Фока и методом теории функционала плотности
- MOLDEN
| Азулен a.pdb(azulene.mol) | Нафталин n.pdb(napthalene.mol) |
 |
 |
нафталин вполне плоский, а вот структура азулена далеко не плоская, поэтому попробуем различные поля для азулена:
| MMFF94 (94.pdb 94.mol) | MMFF94s (94s.pdb 94s.mol) | UFF (uff.pdb uff.mol) |
 |
 |
 |
Затем файл uff.mol и uff_n.mol перекодировали в mop: babel -imol x.mol -omop x_opt.mop -xk PM6 (где х=azu (получен из uff.mol) или nap (получен из uff_n.mol)) Затем запускали мопак: export PATH=${PATH}:/home/preps/golovin/progs/bin export MOPAC_LICENSE=/home/preps/golovin/progs/bin MOPAC2009.exe x_opt.mop
Затем файл x_opt.out перекодировали в inp: babel -imopout x_opt.out -ogamin x_opt.inp
Их можно скачать здесь azu_opt.inp и nap_opt.inp (заголовки уже заменены)
запускаем оптимизацию для этих молекул при помощи GAMESS. На выходе получаем файлы:
azu_opt.log и nap_opt.log
использованный базис - N31
Для рассчета энергии методом Хатри-Фока в начало файлов *.int добавляем такие строчки:
$CONTRL COORD=CART UNITS=ANGS SCFTYP=RHF RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATA
получаем такие файлы azu_hf.inp (азулен) и nap_hf.inp (нафталин)
После gms получаются файлы azu_hf.log (азулен) и nap_hf.log (нафталин)
Для метода теории функционала плотности добавить в начало *.int такие строчки.
$CONTRL COORD=CART UNITS=ANGS dfttyp=b3lyp RUNTYP=ENERGY $END $BASIS GBASIS=N31 NGAUSS=6 POLAR=POPN31 NDFUNC=1 $END $GUESS GUESS=HUCKEL $END $system mwords=2 $end $DATA
получаем такие файлы azu_tf.inp (азулен) и nap_tf.inp (нафталин)
После gms получаются файлы azu_tf.log (азулен) и nap_tf.log (нафталин)
Нашел в log файлах расчёта энергии строчку с "TOTAL ENERGY = " и заполнил таблицу:
| Вещество | Хартри-Фок | DFT |
| Naphthalene | -383.3546611998 | -385.6400108661 |
| Azulene | -383.2824690219 | -385.5857491456 |
| Δ, Hartree | 0,0721921779 | 0,0542617205 |
| Δ, kCal/mol | 45,3 | 34,05 |
Из эксперимента известно, что энергия изомеризации нафталина в азулен составляет 35.3±2.2 kCal/mol. К этому значению приближаются результаты DFT, следовательно, этот метод лучше.
Запускаем X-ming->X-launch через список программ windows. Выбираем удобное расположение окон. В поле Display number установливаем значение 9999. В следующем экране wizard оставляем отметку напротив no client, Next->Next->Finish Запускаем molden.exe. Загружаем файл с результатом DFT, кнопка Read. Переходим в режим плотности ("Dens. Mode"). Выбираем HOMO орбиталь (HOMO-highest occupied molecular orbital ). Строим поверхность для этой орбитали (кнопка Space) со значением контура 0.05. Появляются красно-желтые контуры, переходим в режим OpenGL с помощью кнопки с квадратиками правее кнопки Euclid. Выбираем расположение молекулы и сохраняем изображение. Делаем тоже самое для LUMO (lowest unoccupied molecular orbital)
| HOMO | LOMO | |
| Азулен | 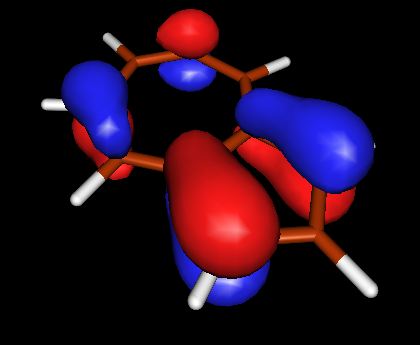 |
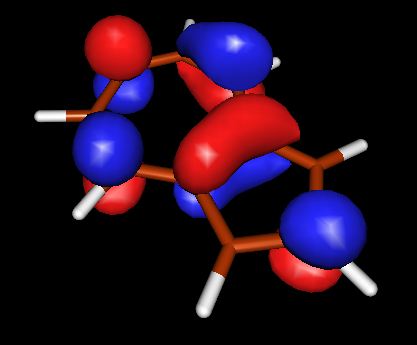 |
| Нафталин | 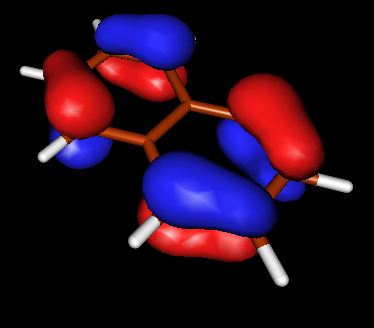 |
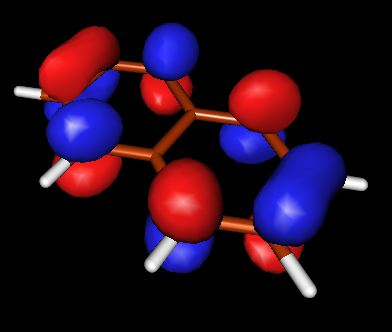 |
из выше приведенных схем видно, что в случае HOMO для азулена и нафталина картины очень похожи, плотность в основном распределена между двумя соседними атомами углерода. В случае LOMO, плотность в основном сконцентрирована на единичных атомах. Стоит отметить, что ни в HOMO, ни в LOMO для нафталина нет плотности, распределенной между атомами С, которые объединяют два бензольных кольца (С-С мостик), в азулене - она есть.
©Анисенко Андрей