Вычисление параметров для молекулярной механики
- C-C связь оптимизированная структура этана в виде Z-matrix:
- Валентные углы
- Торсионные углы
$DATA eth C1 C C 1 cc H 2 ch 1 cchv H 2 ch 1 cch 3 d1 0 H 2 ch 1 cch 3 d2 0 H 1 ch 2 cch 3 d3 0 H 1 ch 2 cch 5 d3 0 H 1 chv 2 cch 4 d3 0 cc=1.52986 ch=1.08439 chv=1.08439 cch=111.200 cchv=111.200 d1=120 d2=-120 d3=180 $END
et.inp - файл загатовка для размножения
make_b.bash - исправленный скрипт (стартовая длинна соответствует файлу et.inp)
после запуска скрипта получили 21 inp файл, в каждом из которых длинна связи С-С -различна (смотрим здесь)
make_bend.bash - конечный вариант скрипта, позволяющий записывать энергию и длину связи с-с.
после запуска скрипта получаем набор значений (длина С-С, энергия):
1.32986 -79.7339315289 1.34986 -79.7406173737 1.36986 -79.7462868578 1.38986 -79.7510331813 1.40986 -79.7549412921 1.42986 -79.7580886263 1.44986 -79.7605457819 1.46986 -79.7623771323 1.48986 -79.7636413832 1.50986 -79.7643920816 1.52986 -79.7646780790 1.54986 -79.7645439543 1.56986 -79.7640303992 1.58986 -79.7631745699 1.60986 -79.7620104053 1.62986 -79.7605689179 1.64986 -79.7588784564 1.66986 -79.7569649422 1.68986 -79.7548520851 1.70986 -79.7525615748 1.72986 -79.7501132582
перенаправим его в bond
ниже приведена зависимость энергии от длинны связи, аппроксимированная f(x)=a+k(x-b)^2
(параметры аппроксимирующей функции:
a=-79.7652+/-0.0004522 k=0.563608+/-0.02335 b=1.55432+/-0.002455
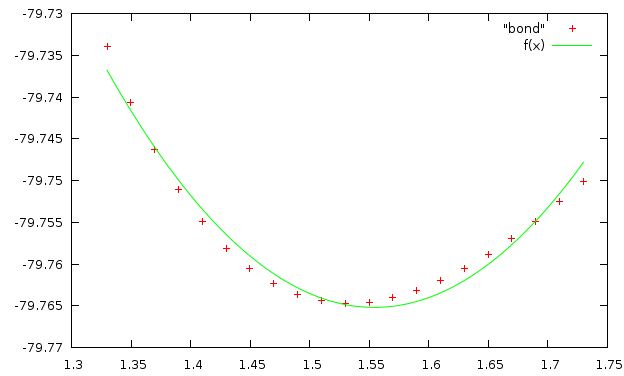
аппроксимирующая кривая и значения плохо совпадают. возможная причина - необходимо использовать более сложную функцию для аппроксимации
Аналогично проводим операции и для валентного угла HCC. Его значения изменяем от 109,2 до 113,2 a.bash - скрипт, выдающий пары валентный угол и энергию молекулы
angles - результат работы скрипта ниже приведена зависимость энергии от длинны связи, аппроксимированная f(x)=a+k(x-b)^2
(параметры аппроксимирующей функции:
a=-79.7647+/-1.21e-08 k=0.563608+/-6.229e-09 b=1.55432+/-9.954e-05
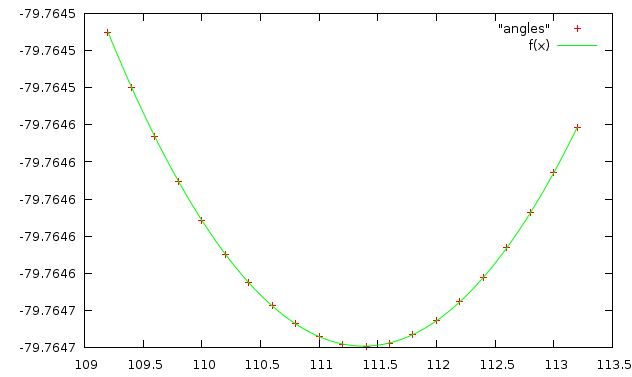
в данном случае эта функция аппроксимировала гораздо лучше.
Аналогично проводим операции и для торсионного угла d3. Его значения изменяем от -180 до 180 с шагом 12 t.bash - скрипт, выдающий пары торсионный угол и энергию молекулы
torsion - результат работы скрипта ниже приведена зависимость энергии от величины торсионного угла d3
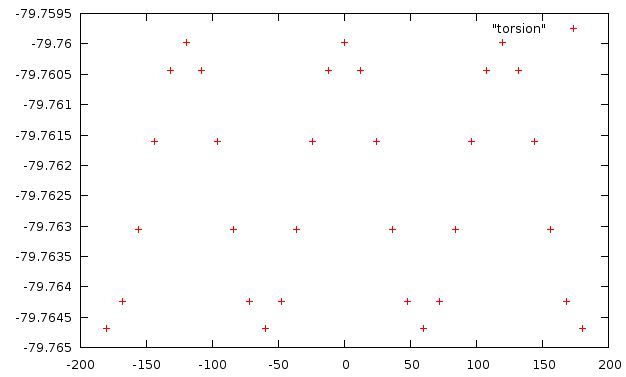
из графика видно, что количество минимумов равно 3 (точки -180 и 180 - одно и то же)
©Анисенко Андрей