Изучение работы методов контроля температуры в GROMACS
- Подготовка файлов
- Анализ результатов
- Метод Берендсена
- Метод Андерсена
- Метод стохастической молекулярной динамики
- Метод Нуза-Хувера
- Метод "Velocity rescale"
Начнем с того, что подготовим файл координат и файл топологии. На прошлом занятии мы использовали gro файл с 38 молекулами этана. Создадим индекс файл в котором будет группа из одной молекулы этана.
make_ndx -f box_38.gro -o 1.ndx После запуска команды появляется приглашение к вводу. Сначала ознакомимся с помощью, нажав "h" + enter. Задаем остаток номер 1 (r1+enter). Теперь создадим gro файл с одной молекулой и зададим ячейку. При запуске ediconf выбираем номер соответствующей группы из одной молекулы.
editconf -f box_38.gro -o et1.gro -n 1.ndx
#зададим ячейку и расположим молекулу по центру ячейку
editconf -f et1.gro -o et.gro -d 2 -c
Исправим файл топологии et.top из прошлого задания. Т.к. на прошлом практикуме для разных атомов углерода мы получили разные заряды, попробуем исправить входной файл Mol_red1.p2n (текст исходного файла приведен ниже).
REMARK REMARK TITLE MOLECULE REMARK CHARGE-VALUE 0 REMARK MULTIPLICITY-VALUE 1 REMARK ATOM 1 CT1 LIG 1 0.000 0.000 -0.765 C ATOM 2 H1 LIG 1 -1.011 0.000 -1.157 H ATOM 3 H1 LIG 1 0.506 0.876 -1.157 H ATOM 4 H1 LIG 1 0.506 -0.876 -1.157 H ATOM 5 CT2 LIG 1 0.000 0.000 0.765 C ATOM 6 H2 LIG 1 1.011 0.000 1.157 H ATOM 7 H2 LIG 1 -0.506 0.876 1.157 H ATOM 8 H2 LIG 1 -0.506 -0.876 1.157 H CONECT 1 2 3 4 5 CONECT 2 1 CONECT 3 1 CONECT 4 1 CONECT 5 1 6 7 8 CONECT 6 5 CONECT 7 5 CONECT 8 5 END
Для того, чтобы скрипты RED распозновали правильно одинаковы атомы этана (С атомы), необходимо их переименовать из СТ1 и СТ2 и назвать одинаково.
В разделе [ molecules ] измените количество молекул этана.
конечный вариант et.top
Даны 5 файлов с разными параметрами контроля температуры:
be.mdp- метод Берендсена для контроля температуры.
vr.mdp- метод "Velocity rescale" для контроля температуры.
nh.mdp- метод Нуза-Хувера для контроля температуры.
an.mdp- метод Андерсена для контроля температуры.
sd.mdp- метод стохастической молекулярной динамики.
Ниже приведен текст скрипта, позволяющий одновременно работать со всеми выше описанными файлами:
for i in { an , be , nh , sd , vr };do
# grompp -f ${i}.mdp -c et.gro -p et.top -o et_${i}.tpr
# mdrun -deffnm et_${i} -v -nt 1
# trjconv -f et_${i}.trr -s et_${i}.tpr -o et_${i}.pdb
# g_energy -f et_${i}.edr -o et_${i}_en.xvg
g_bond -f et_${i}.trr -s et_${i}.tpr -o bond_${i}.xvg -n b.ndx
done
Для каждой из пяти систем получили pdb файлы для визуализации процессов, протекающих в системе:
et_be.pdb- метод Берендсена для контроля температуры.
et_vr.pdb- метод "Velocity rescale" для контроля температуры.
et_nh.pdb- метод Нуза-Хувера для контроля температуры.
et_an.pdb- метод Андерсена для контроля температуры.
et_sd.pdb- метод стохастической молекулярной динамики.
Немного предварительных выводов:
Сначала молекула этана практически не вращается (наблюдается лишь изменение торсионных углов НССН), постепенно молекул выходит из состояния равновесия и начинает колебаться, постепенно наращивая амплитуду и частоту колебаний. Такое поведение молекул этана трудно представить для системы, находящейся при одной и той же температуре, которую должен обеспечивать наш термостат. Такое поведение молекул с наибольшей вероятностью может быть реализовано в системе с изменением агрегатного состояния (твердый этан при низких температурах медленно нагревается, и через стадию жидкого переходит в газообразный).
Наблюдаются незначительные колебания длин связей (молекула "дрожит"), но сама молекула не изменяет сввоего положения. Данный метод аналогично предыдущему выглядит наименее правдоподобным, а полученная система скорее всего демонстрирует состояние этана при сверхнизких температурах ( в твердом состоянии) (Т.е. в принципе, температура поддерживается на сверхнизком уровне, но возникает вопрос о применение такого термостата для моделирвания...)
Молекула изменяет свое положение с более или менее одинаковыми скоростями и амплитудами. Если просмотреть ролик покадрово, то в любой момент времени молекулы этана представлены заторможенной конформацией (что и логично, т.к. заслоненная конформация энергетически менее выгодна, чем заторможенная) Данный термостат (по предварительной визуальной оценке) может обеспечивать постоянную температуру в системе с хорошей точностью.
В молекуле происходит изменение лишь НССН торсионных углов. длина СС связи не изменяется. Часто можно увидеть заслоненную конформацию этана, что не очень выгодно для системы. Такой термостат подает в систему мало энергии.
Напоминает метод Нуза-Хувера, но молекулу реже можно заметить в заслоненной или близкой к ней конформации. добавляется пространственное вращение всей молекулы
Предварительный вывод: неплохо справились с задачей метод стохастической молекулярной динамики и "Velocity rescale".
Перейдем к более детальному изучению результатов работы. На рисунке ниже приведены зависимотсти энергий (потенциальной (красные крестики) и кинетической (зеленые крестики) от времени (состояния системы)) и распределение длинны связи.
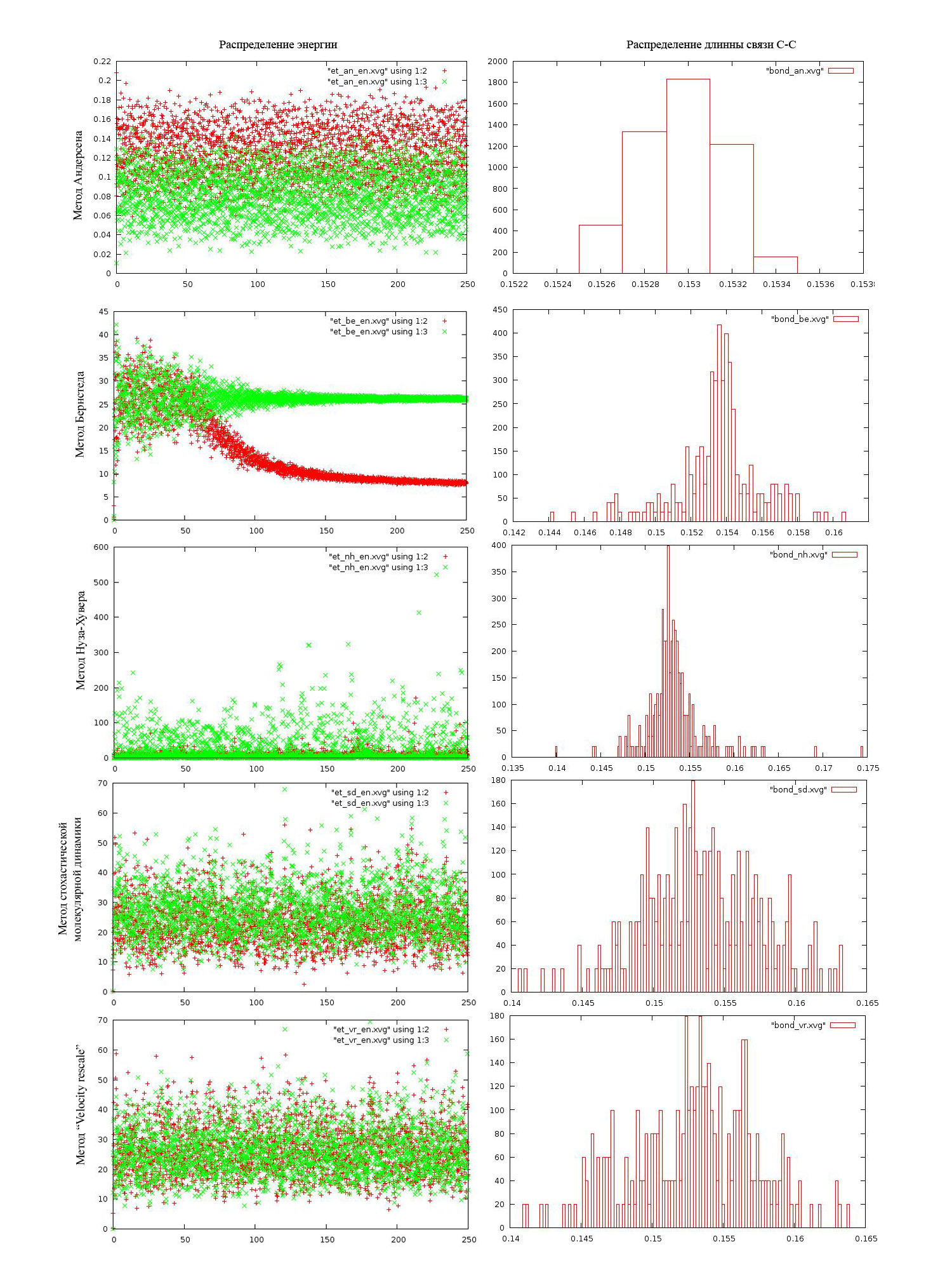
В случае метода Андерсена для системы характерны низкие значения как кинетической так и потенциальной энергии. Длинна связи С-С меняется незначительно.
В случае метода Бренстеда картина чуть лучше, но через некоторое время потенциальная энергия опускается до нуля, а кинетическая "концентрируется" около одного значения (происходит уменьшение разброса)
В случае метода Нуза-Хувера и потенциальная, и кинетическая энергии сконцентрированны около от 0 до 100 (хотя есть большое количество точек, кинетическая энергия которых принимает значительно большие значения). Из распределения длинн связей видим, что С-С связь практически не колеблется
Все выше описанное не совпадает с реально возможными системами. в приведенных системах либо очень низкие температуры, либо они просто не могут существовать. Зато, при использовании методов стохастической молекулярной динамики и "Velocity rescale" получили неплохие результаты:есть некий разброс энергий около среднего значения, причем чем больше отклонения энергии системы от среднего, тем реже мы встречаем такие состояния (т.е. зависимости согласуются с распределением Больцмана:
ni = A wie-ei/kT (1)
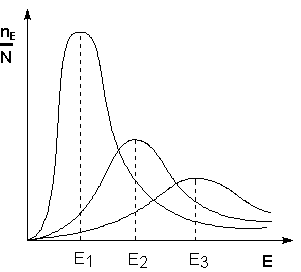
), плюс распределение длинн С-С свзяей имеет разумный вид. Наши предположения о этих методах подтвердились (они действительно хорошо справились с задачей поддержания температуры системы).
©Анисенко Андрей