Гомологичное моделирование комплекса белка с лигандом
- Подготовка файлов При помощи программы muscle, установленной на кодомо, создал выравнивание своего белка (lys_antmy.fasta) c лизоцимом из радужной форели. Выравнивание было импортировано в GeneDoc и сохранено в формате PIR (1out.pir)
Для дальнейшей работы модифицируем файл выравнивание:
1. Переименовываем последоваельность в файле выравнивания, как описано в примере:
Было |
Стало |
>P1;uniprot|P37712|LYSC_CAMDR |
>P1;seq |
>P1;1LMP|PDBID|CHAIN|SEQUENCE |
>P1;1lmp |
2.После имени моделируемого белка добавляем строчку: sequence:ХХХХХ::::::: 0.00: 0.00 (входные параметры для modeller)
После имени белка-образца: structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00 (содержит информацию о файле со структурой белка, номер первого и последнего остатка, идентификатор цепи)
3. В конце каждой последовательности добавляем /. (/-конец цепи белка, . -количество лигандов).
2out.pir - конечный вариант выравнивания
4. Модификация файла со струтурой: удаляем молекулы воды, всем атомам лиганда присваиваем один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицируйем имена атомов каждого остатка, добавив в конец буквы A, B, C (смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д).
5. Создадим управляющий скрипт.
Для этого вначале проанализируем одородные связи в молекуле 1LMP, которые обеспечивают удержание лиганда в структуре белка.
Ниже приведена структура белка с лигандом и водородными связями (обозначены желтым пунктиром):
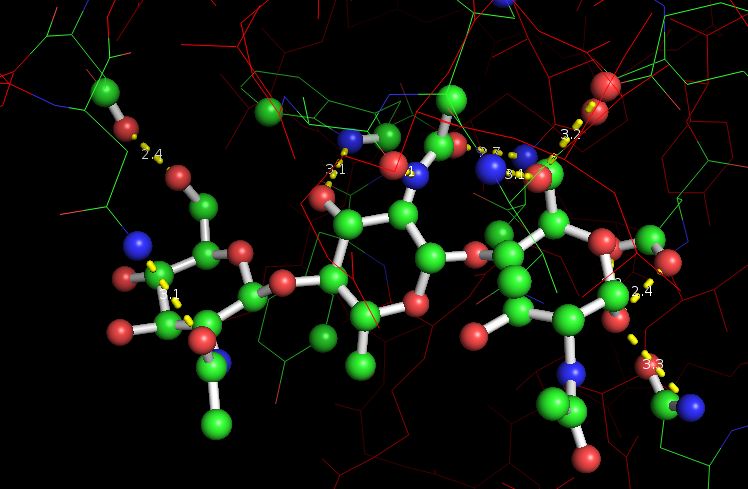 |
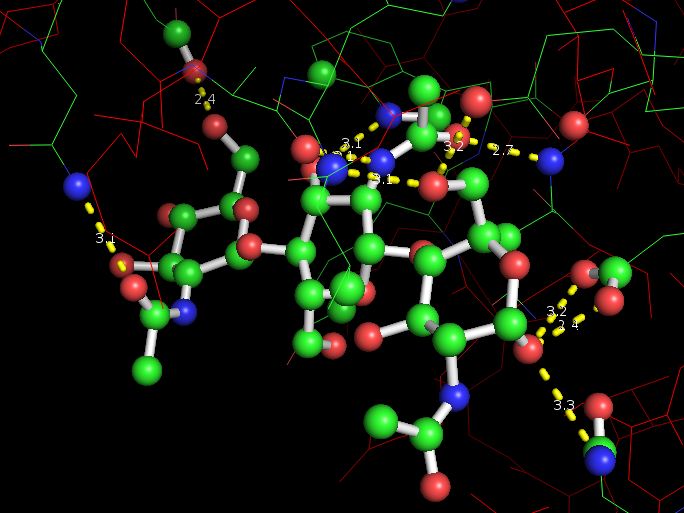
|
на схеме, приведенной ниже, мы видим остатки и атомы в их составе, которые формируют водородные связи.
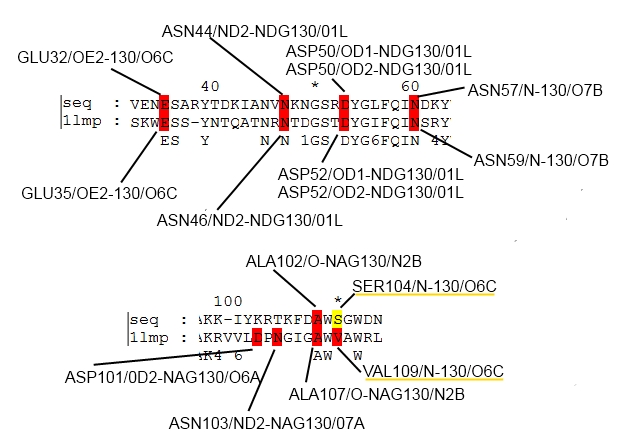
над верхней последовательностью (seq) подписаны контакты в будущей модели. Видим, что в будущей моделе будут отсутсвовать две водородные связи, образованные аминокислотами с номерами 101 и 103. видимо данные водородные связи наименее важны для связывания лиганда. Также в выравнивании 109VAL последовательности 1lmp заменен на 104SER. Но валин формировал водородную связзь с лигандом посредством амино группы в составе пептидной связи. Таким образом при моделировании структуры нового белка бы запустить скрипт два раза:
1)не учитывая контакт SER104/N-130/O6C,
2)учитывая контакт SER104/N-130/O6C.
(Но мы обойдемся без этого, т.к. того количества водородных связей, которое мы используем уже достаточно для точного построения структуры белка (минимальное количество водородных свзяей для правильной пространственной фиксации лиганда = 3))
В результате работы скрипта lys_antmy.py получили следующие модели:
seq1.pdb
seq2.pdb
seq3.pdb
seq4.pdb
seq5.pdb
6. Анализ результатов
Для оценки качества структур воспользуемся средствами, предоставляемыми ресурсом WHAT IF.
в нем собран огромный пакет для работы с различными струкурами. Например, для проверки правильности моделей есть специальный пакет алгоритмов Structure validation. Ниже в таблице приведены значения для исследуемых параметров (1, 2, 3 .. - номер модели, 0 - исходная структура).
Будим считать лучшей моделью ту стурктуру, параметры которой наиболее близки к параметрам исходной pdb структуры
| № модели | RMS Z-score for bond lengths | Ramachandran Z-score | RMS Z-score for bond angles |
| 1 | 1.027 | -2.173 | 1.422 |
| 2 | 1.029 | -1.915 | 1.437 |
| 3 | 1.034 | -2.118 | 1.505 |
| 4 | 1.029 | -1.943 | 1.385 |
| 5 | 1.017 | -1.660 | 1.439 |
| 0 | 0.839 | -0.965 | 1.752 |
Первые два параметра ближе всего к показателям нулевой модели (исходная структура) в случае пятой модели. далее свою работу удем проводить именно с ней
©Анисенко Андрей