Задание 1. Объяснение странностей в PDB файлах.
7GPB
PDB: 7GPB
Год расшифорвки: 1991 (выход статьи), 1990 (внесение в PDB). Последнее редактирование произведено в 2011 году, но относилось к добавлению альтернативного названия лиганда и обновлению версии (REVDAT).
Разрешение: 2.90 Å
Странность.
В данной записи интересны остатки триптофана. Кольца этой аминокиcлоты образуют сопряженную пи-систему, то есть p-атомные орбитали перекрываются и образуют две плоскости своими зонами перекрытия. Из-за этого атомы (по сути - центры сгущения электронной плотности) колец триптофана должны лежать в одной плоскости. Выход хотя бы одного атома из пи-системы требует разрыва этой системы и больших затрат энергии.
При обработке данных авторы 7GPB не учли это химическое свойство для нескольких колец и получили абсурдные результаты, один из которых показан на рис.1.
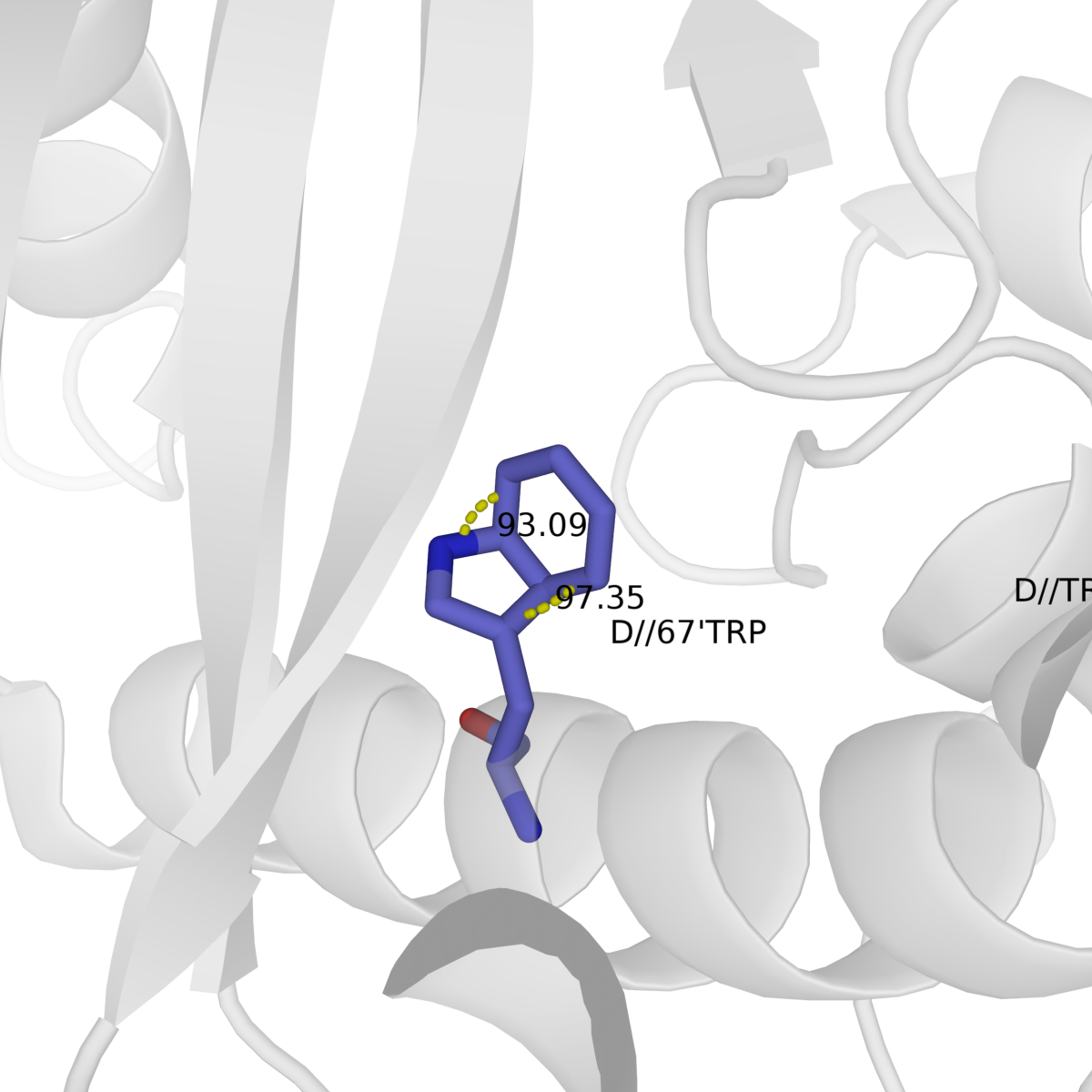
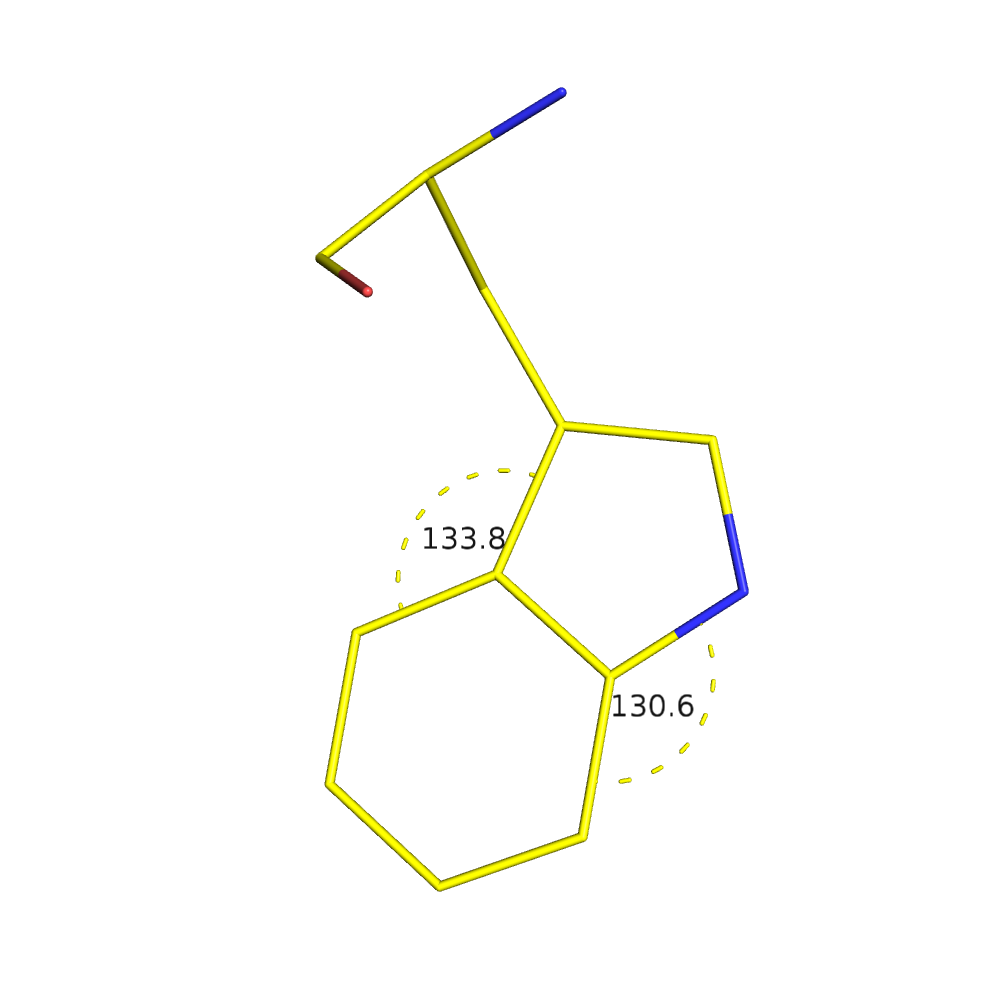
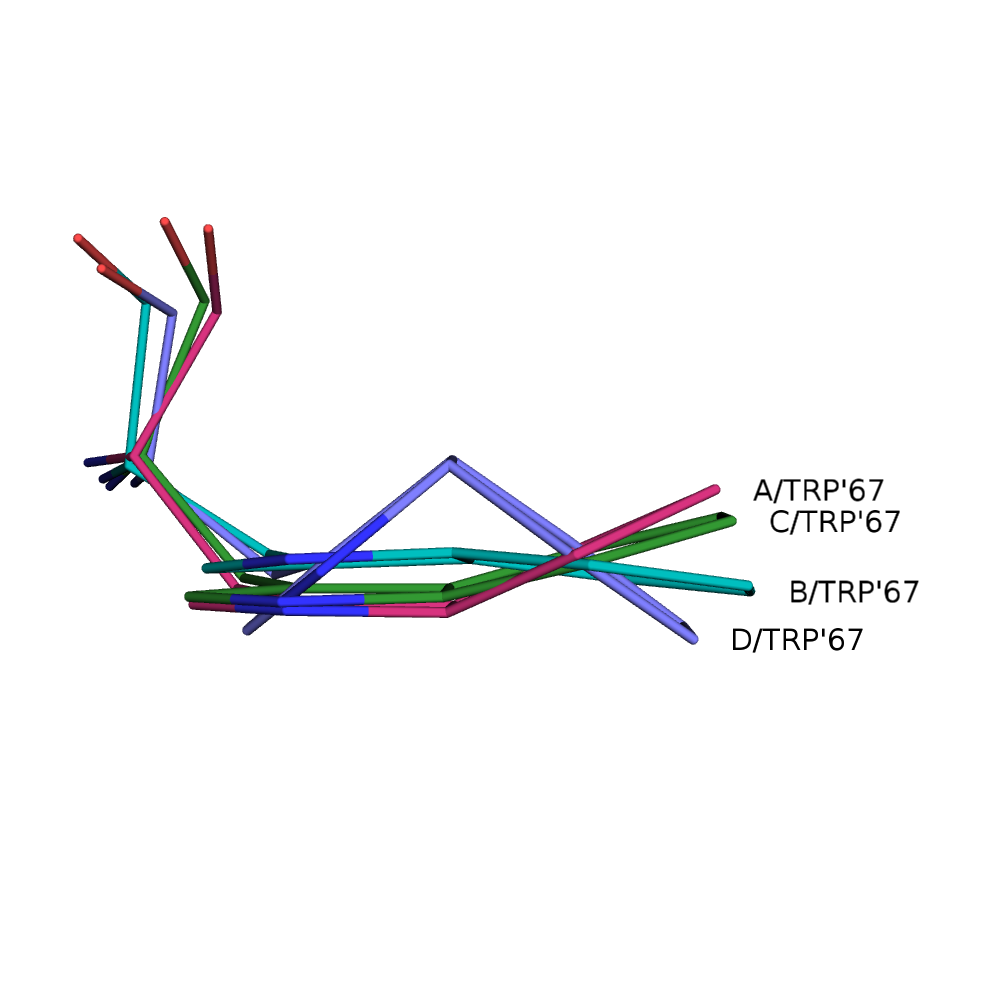
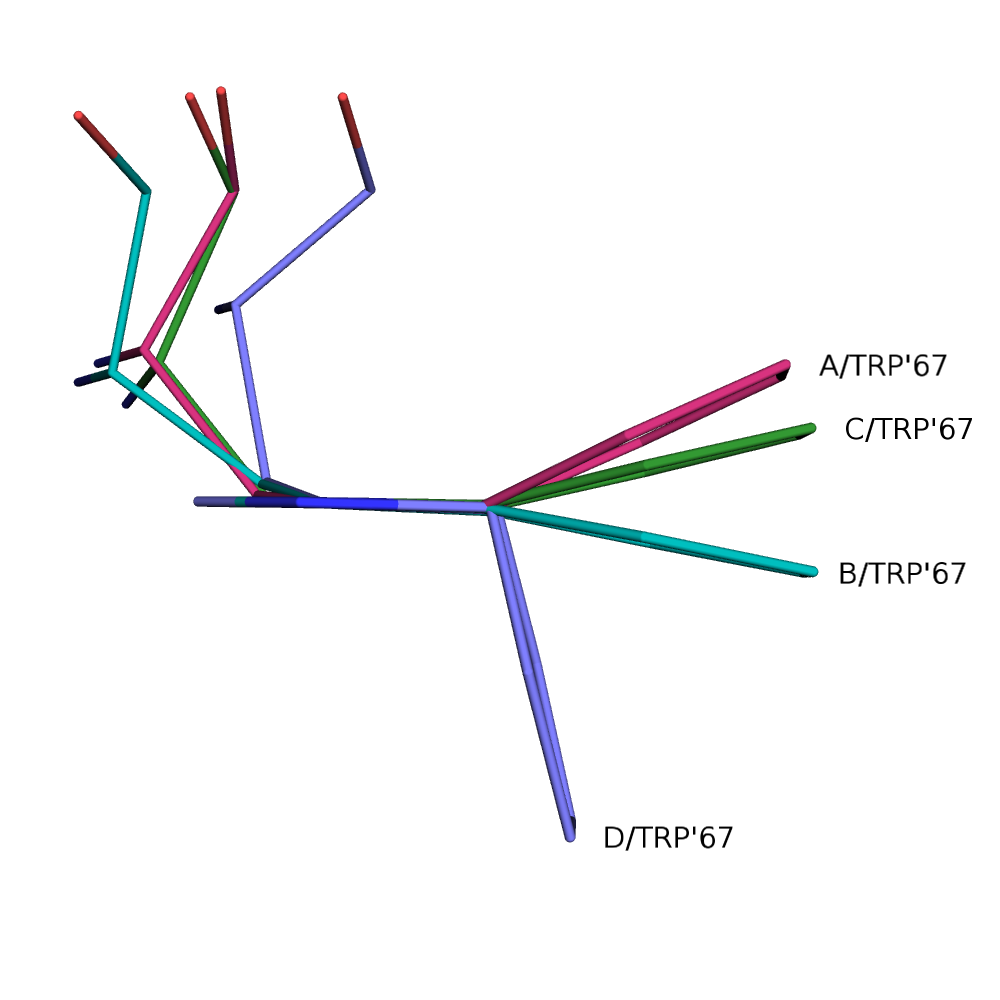
Вообще, можно взять триптофаны с теми же номерами из всех цепей белка 7GPB, выровнять целиком или по отдельным атомам и посмотреть, как сильно их конформация отличается от нормы - см. рис. 3.
Возможное объяснение.
Скорее всего, авторы использовали некачественный софт, который не предусматривал все особенности химического строения аминокислот белков.
Но ошибки могли бы не проявиться столь резко, если температурные факторы атомов не были такими большими - более 40, вплоть до 60. Это означает, что электронная плотность соответствующих атомов "размазана" в пределах 6-8 ангстрем. Возможно, стоило бы учесть альтернативные конформации на месте таких атомов. С другой стороны, возможно, белок просто плохо кристаллизовался и данные положения не могут быть определены более точно. К сожалению, данных экспериментальных структурных факторов нет в открытом доступе, поэтому более четкое объяснение предложить трудно.
В любом случае, явно то, что из всех вариантов структур химически неверные лучшим образом подходят под экспериментальные данные (по параметрам используемых авторами программ).
5RXN
PDB: 5RXN
Год расшифорвки: 1984 (внесение в PDB). Последнее редактирование произведено в 2009 году, но относилось к обновлению версии (REVDAT).
Разрешение: 1.20 Å
Странность.
Всего 2 аминокислоты в составе белков обладают хиральностью при бета-атоме углерода: треонин и изолейцин. На рисунке ниже приводятся изомеры их L-форм (D-изомеры здесь не рассматриваю). В природе встречается только один энантиомер из четырех возможных для аминокислоты с двумя хиральными центрами.

Авторы записи 5RXN не учли хиральность бета-атома углерода треонина. Всего в их белке 3 этих аминокислоты.
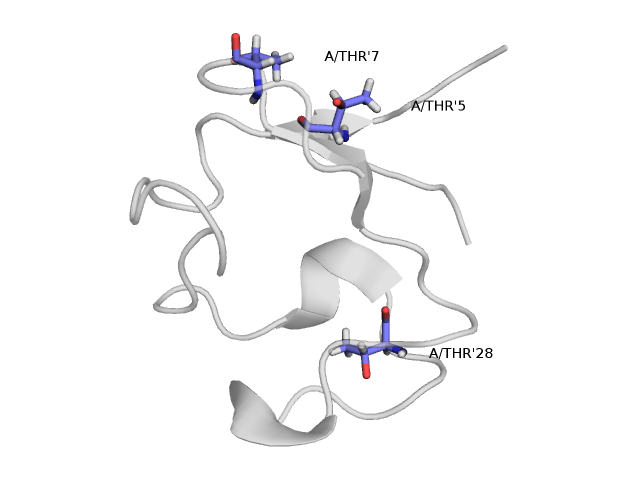
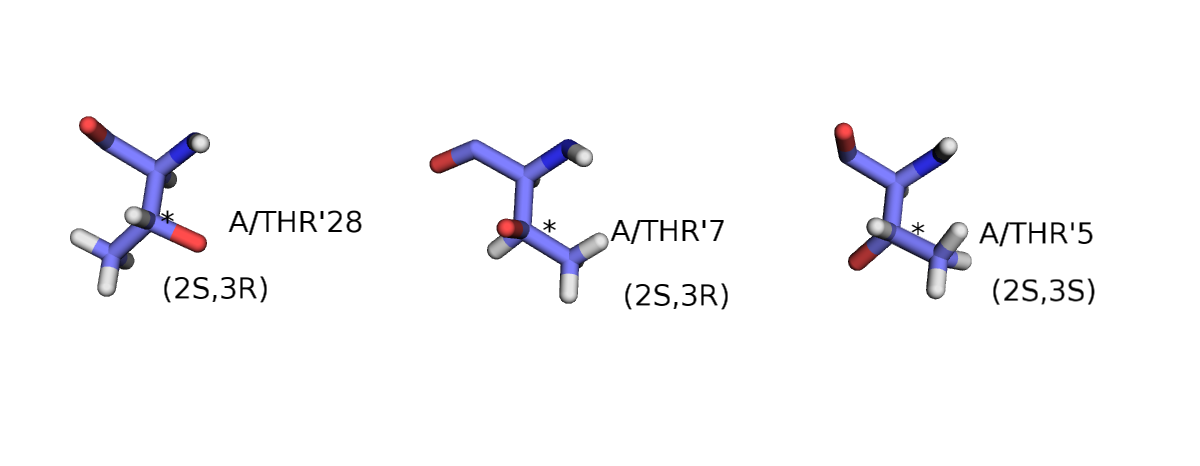
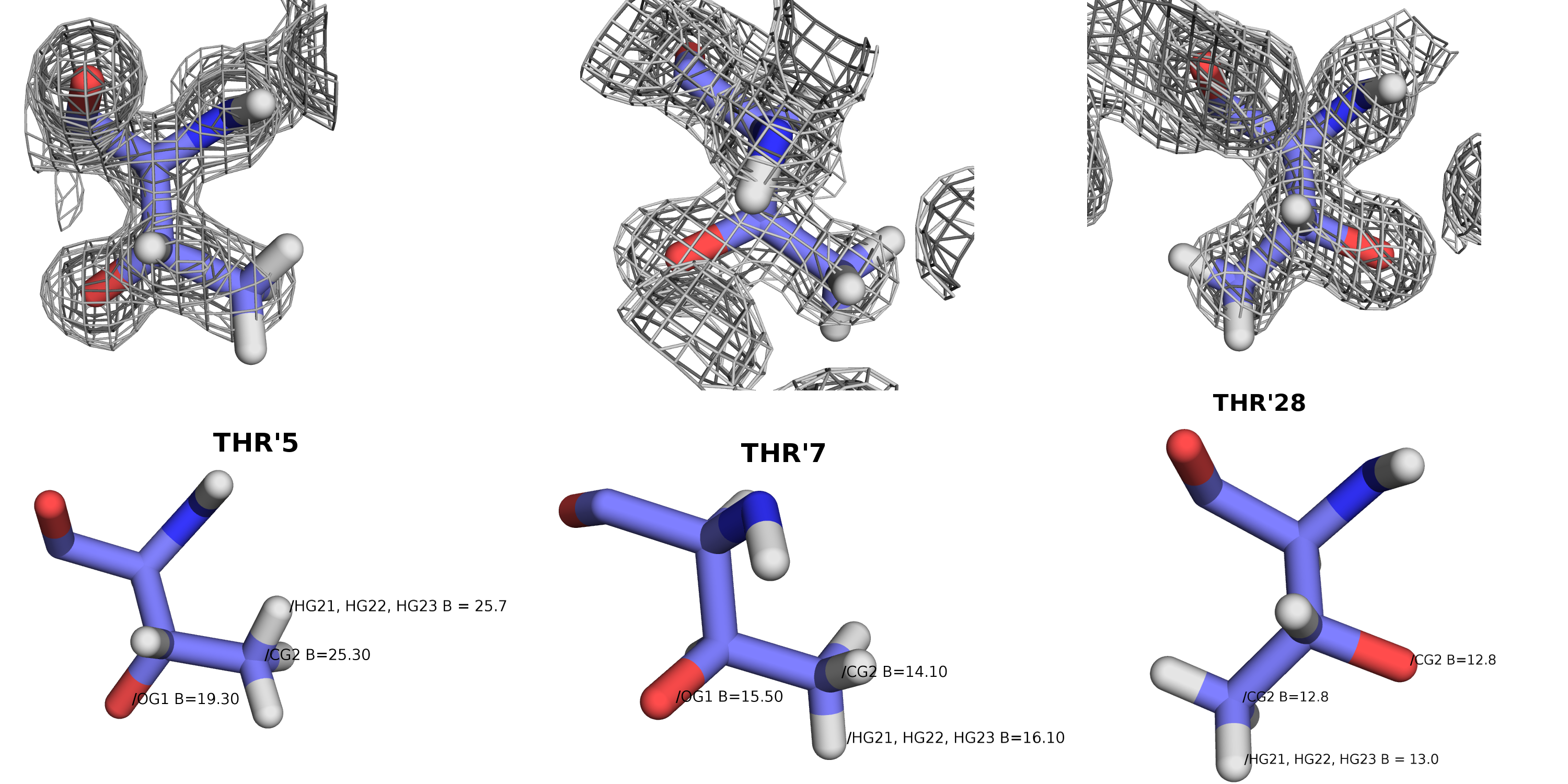
При детальном рассмотрении видно, что только два треонина из трех соответсвует реальному энантиомеру, а THR'5 - ошибочный, имеет конфигурацию (2S,3S), не является природной аминокислотой.
Возможное объяснение.
Положение атомов радикала THR'5 менее определено: температурные факторы больше таковых атомов нормальных треонинов.
Вообще, при рассмотрении электронной плотности представляется вполне возможным, что программа, не учитывающая конформацию CB-атома треонина, неправильно расположила атом OG1 и метильную группу CG2+HG2*. Пик кислорода оказался сравним с размазанной суммой пиков C+3H, и по каким-то причинам они были перепутаны. А именно перестановка этих двух групп определяет энантиомер.
1GT0
PDB: 1GT0
Год расшифровки: 2003 (публикация статьи), 2002 (внесение в PDB). Последние редактирования производились в 2012 году, относились к дополнению описания, синхронизации с базой данных последовательности, а также проверке геометрии молекулы.
Разрешение: 2.60 Å
Странность.
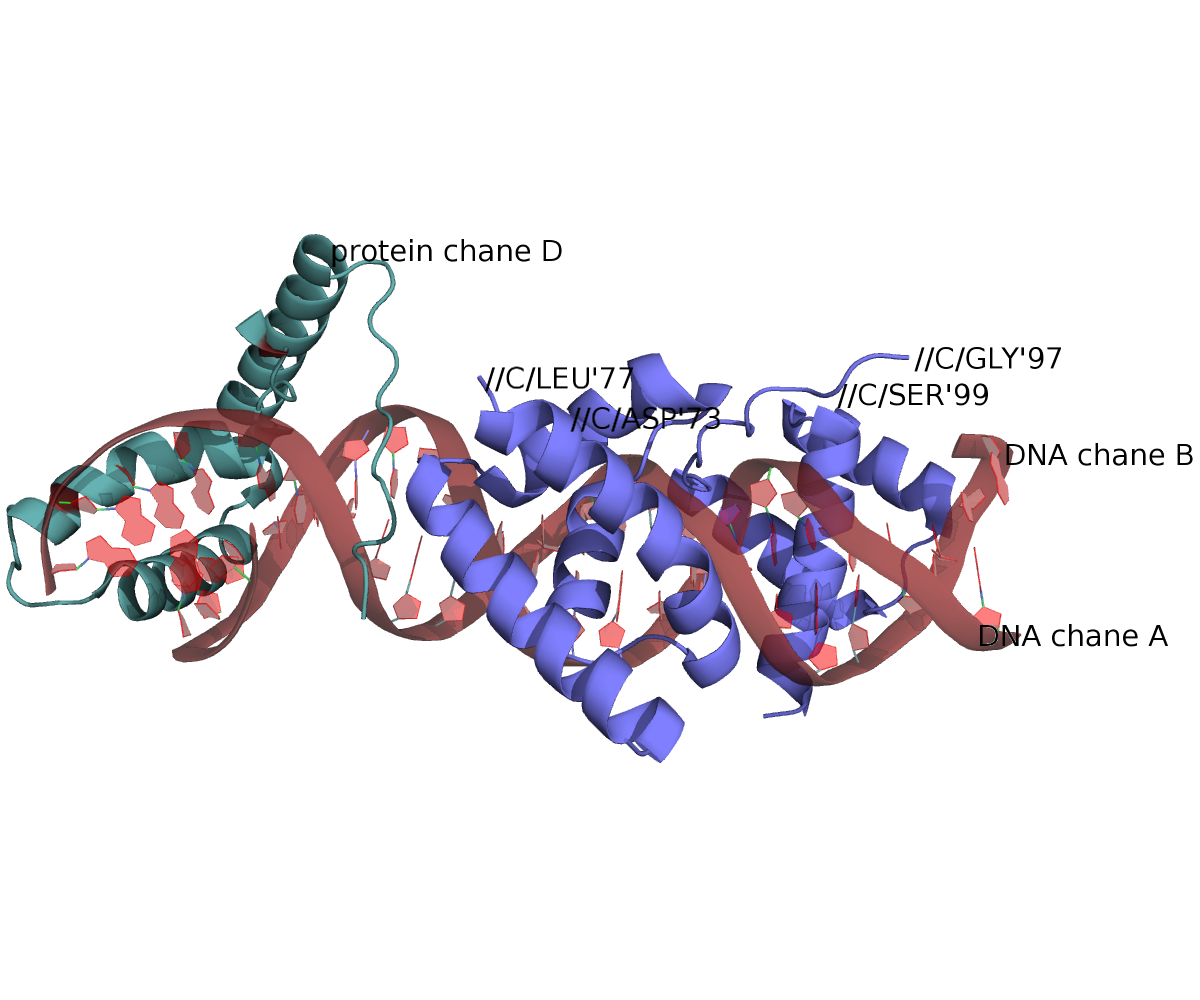
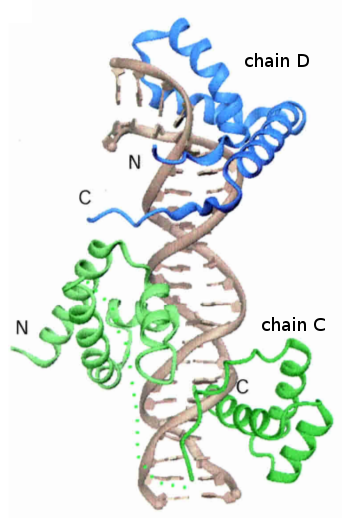
В цепи C представленной записи отсутствуют остатки с 78 по 96. Это можно понять не только при просмотре файла в PyMol, но и прописано в самом файле в разделе REMARK, параграф MISSING RESIDUES. Там же прописано, что не были кристаллизованы еще N- и C-концевые остатки цепи C. К сожалению, большей информации в файле не приводится. В статье авторов об этой структуре также не упоминается об отсутствующих фрагментах. Вообще же, статья авторов была посвящена ДНК-белковым комплексам, для исследования были использованы мутантные ДНК-связывающие домены: POU-домен транскрипционного фактора 1 (150 остатков, цепь C) и HMG-домен транскрипционного фактора SOX-2 (79 остатков, цепь D).
Возможное объяснение.
Первое предположение - что данный участок цепи очень подвижный и плохо закристаллизовался. Действительно, в файле PDB сразу бросается в глаза высокие температурные факторы - около 50 в среднем, порядка 90 у атомов соседних к фрагменту аминокислот. Возможно, что этот фрагмент был настолько подвижен и неточно разрешим, что авторы его исключили из структуры. К счастью, в открытом доступе имеется файл структурных факторов, по которым сервер EDS может восстановить электронную плотность. Если предположение верно, на месте удаленного фрагмента можно было бы наблюдать сильно размазанную электронную плотность. Но как можно видеть из рисунков ниже, это не так, на месте фрагмента электронная плотность просто "обрывается", а на месте этого фрагмента лежит альфа-спираль соседнего по ячейке белка.

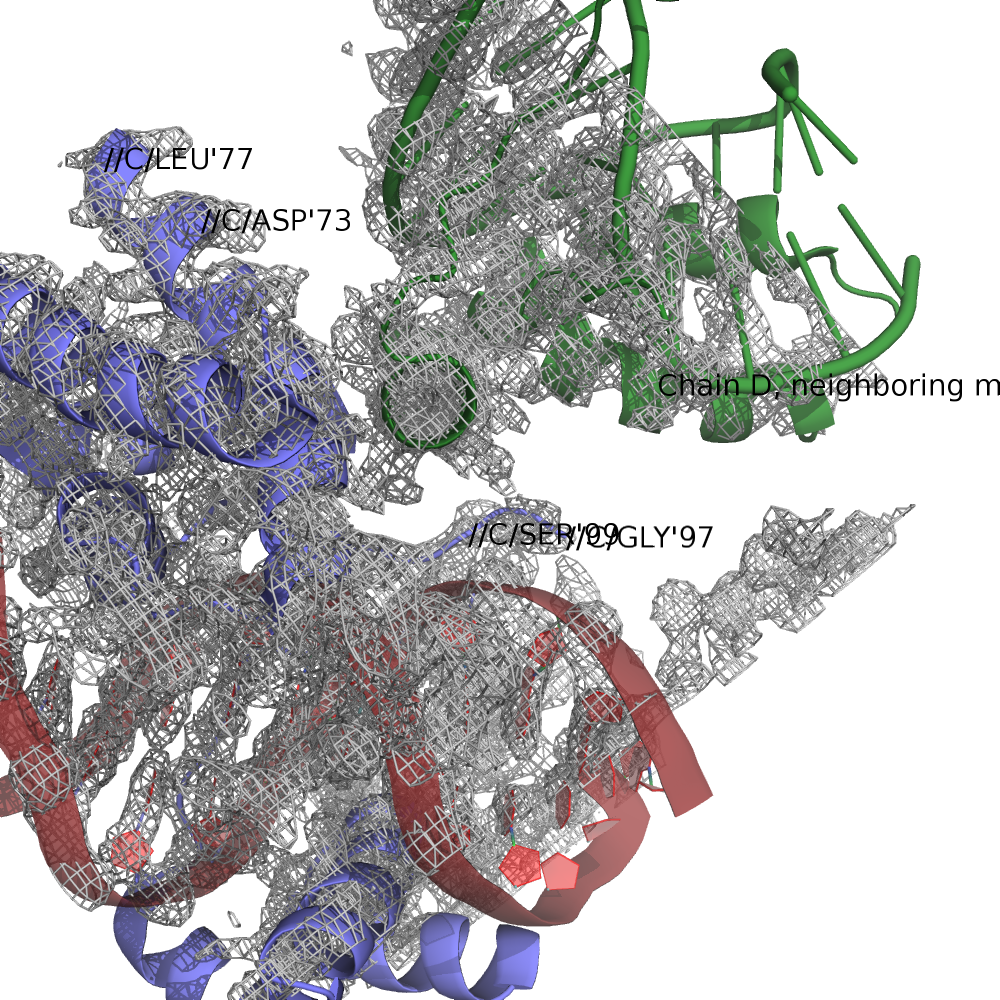
Домен POU (цепь C) в норме состоит из двух ДНК-связывающих субдоменов и линкерного участка. Автора структуры утверждают в своей статье (Remenyi et al. 2003), что в работах с POU до них тоже наблюдался "невидимый" участок линкера (Klemm et al. 1994; Remenyi et al. 2001), хотя и был короче. В любом случае, для поиска ДНК-белковых контактов этот линкере играет важной роли.
Задание 2. Исследование взаимосвязи между разрешением структуры, температурным фактором и коэффициентом заполнения.
Выбор структур
В предложенном задании требуется выбрать одну структуру с очень хорошим разрешением, менее 1 ангстрема, и одну структуру со средним разрешением 2.0 - 2.5 ангстрем.
Иллюстрация выбора студента при заданных ограничениях приведена на графике ниже.
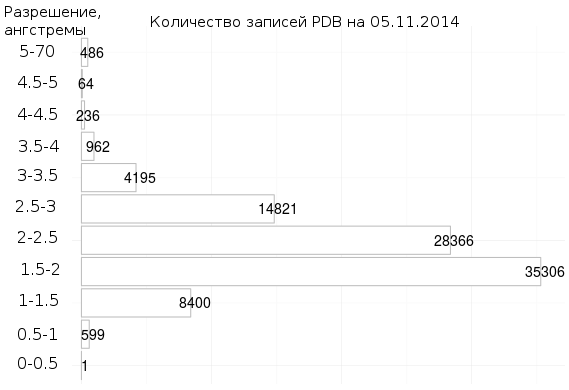
Чтобы не пропустить важный эффект, лучше наложить дополнительные требования на поиск структур: белки должны быть примерно одинаковые по длине, схожих типов.
Например, под заданные требования хорошо подходят мономеры флуоресцентных белков. Со средним разрешением была выбрана структура GFP 4OGS (2.2 ангстрем, длина 238), с очень хорошим - производное GFP 4Q9W (1.0 ангстрем, длина 220). Для сравнения также выбрана структура GFP 4JFG (3.0 ангстрем, длина 244) - с плохим разрешением. На сервере EDS можно найти файлы структурных факторов и электронную плотность для этих записей.
Исследование связи коэффициента заполнения и разрешения
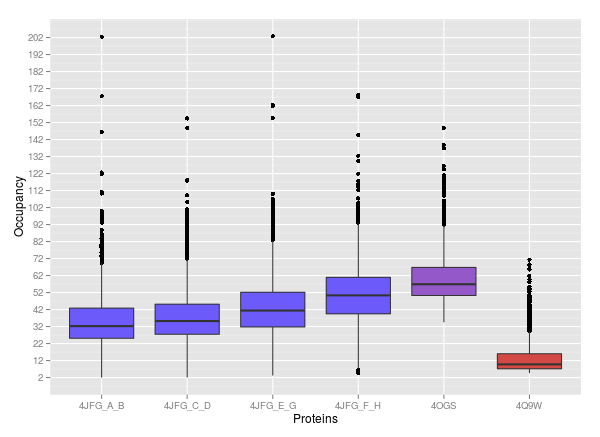
Сравнение коэффициента заполнения для всех трех структур дает ожидаемые результаты: чем лучше разрешение, тем больше позиций с неполным заполнением.
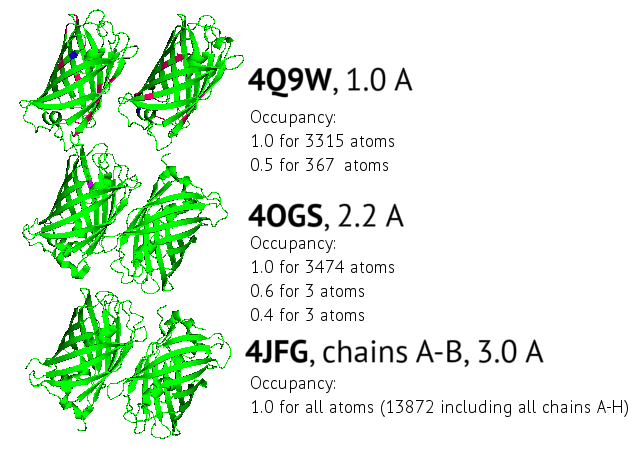
Это объясняется тем, что с лучшим разрешением можно более достоверно различить альтернативные конформации аминокислот.
Исследование связи температурного фактора и разрешения
Сравнение температурных факторов 4Q9W (отличное разрешение) и 4OGS (среднее разрешение) дает ожидаемые результаты: температурный фактор заметно больше для структуры с худшим разрешением. Чем лучше (т.е. меньше по величине) разрешение, тем точнее можно определить положение атомов.
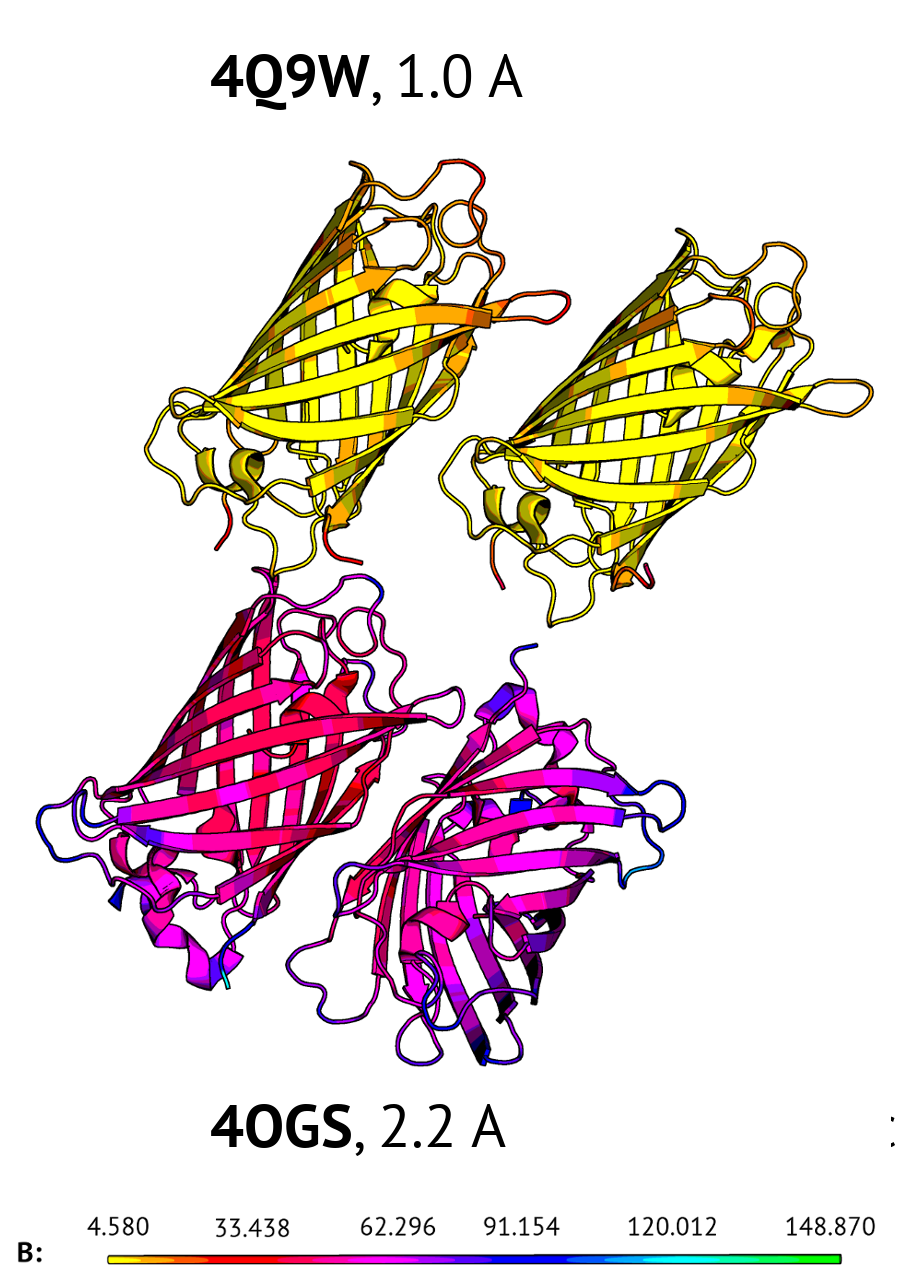
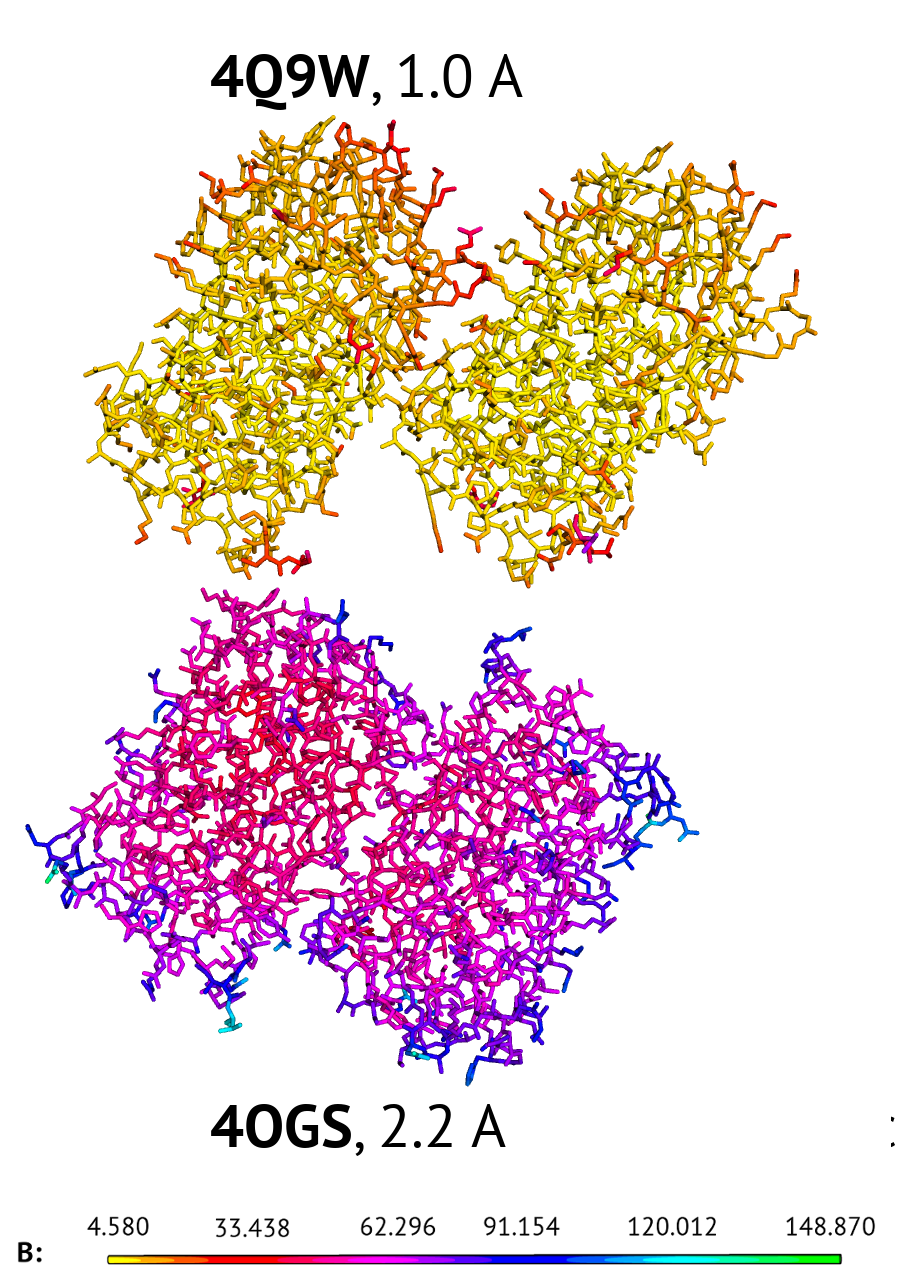
Но рассмотрение структуры с плохим разрешением меняет картину: температурный фактор структуры с плохим разрешением принимает средние значения между средними значениями отличного и нормального разрешения. Это можно увидеть из изображений ниже.
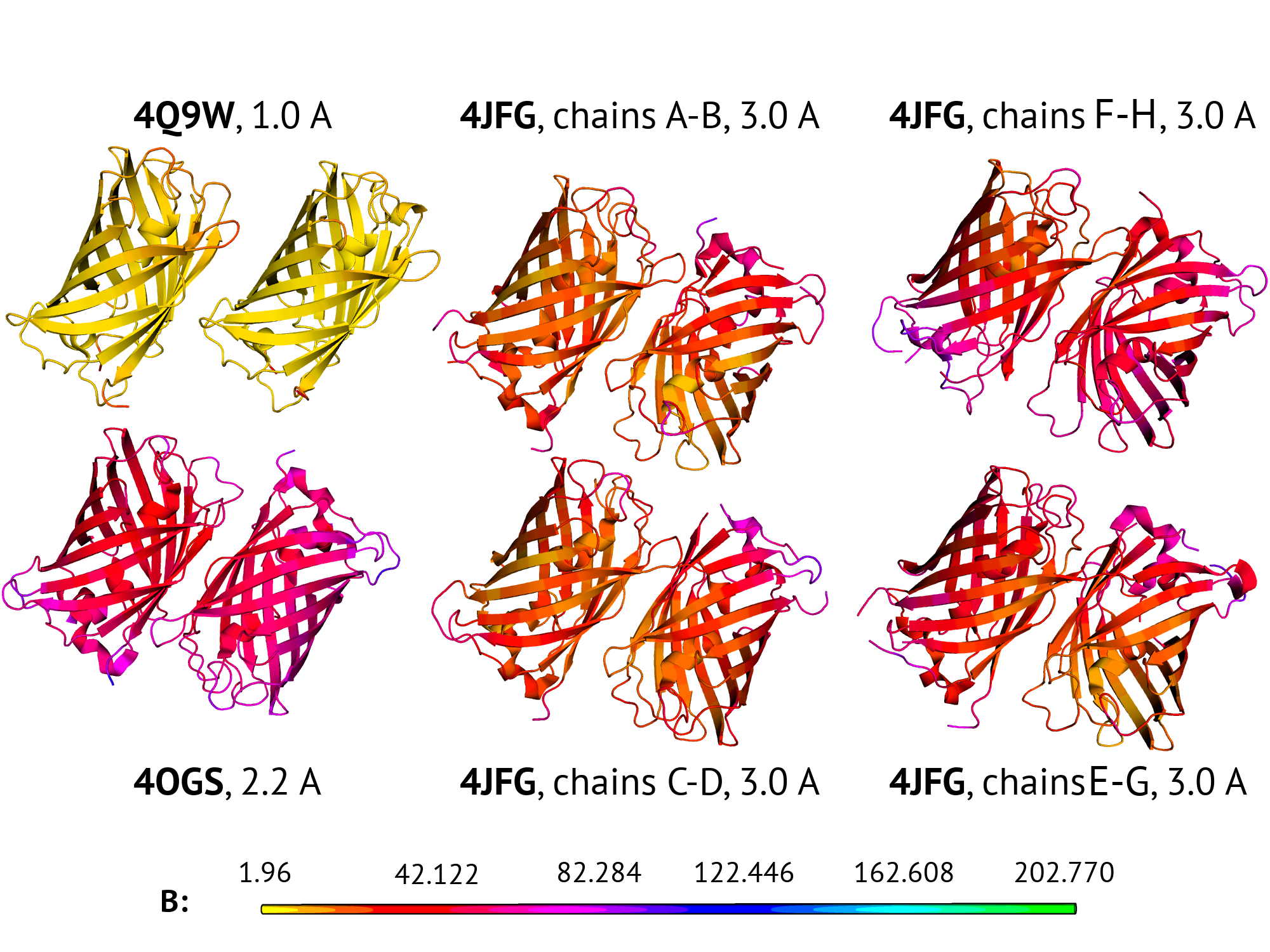
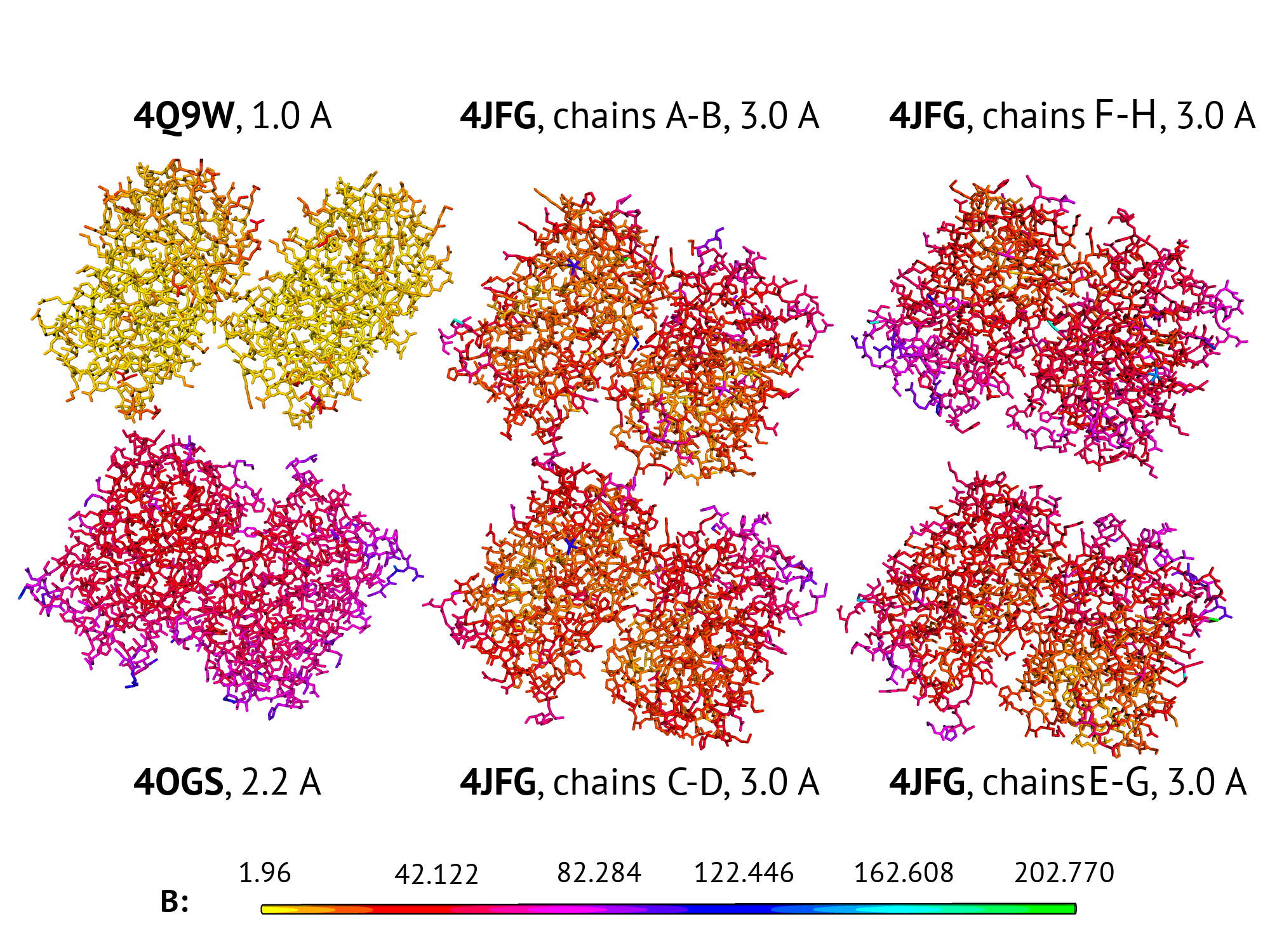
Стоит отметить, что структура 4JFG сама по себе довольно странная. Для начала, на сервере PDB указана длина последовательности 244 и изображены только две цепи G и H; при этом в записи PDB указано уже 8 цепей c суммарной длиной более тысячи аминокислот. Это означает несовпадение асимметрической и биологической единиц. Из статьи авторов (X.Liu et al, 2013) становится понятно, что авторы не гнались за высоким разрешением и качеством структуры: публикация посвящена химическим свойствам разработанного ими варианта GFP. Cама структура в статье упоминается в одном абзаце. Все восемь цепей представляют собой октамер GFP, из которого можно выделить самостоятельные димеры. Для всех четырех димеров средние значения температурного фактора заметно различаются.
Интересно, что как раз в структуре 4JGS можно найти максимальные значения температурного фактора среди всех структур. Возможно, именно эти плохо разрешенные атомы определяют суммарное плохое разрешение структуры. Эти плохо разрешенные атомы - ионы цезия, в котором производилась кристаллизация белка. Информация о назначении этих ионов не была мною найдена: метод получения структуры - молекулярное замещение, не связан с ионами тяжелых металлов. Возможно, условия добавления CsCl просто были достаточными для кристаллизации.
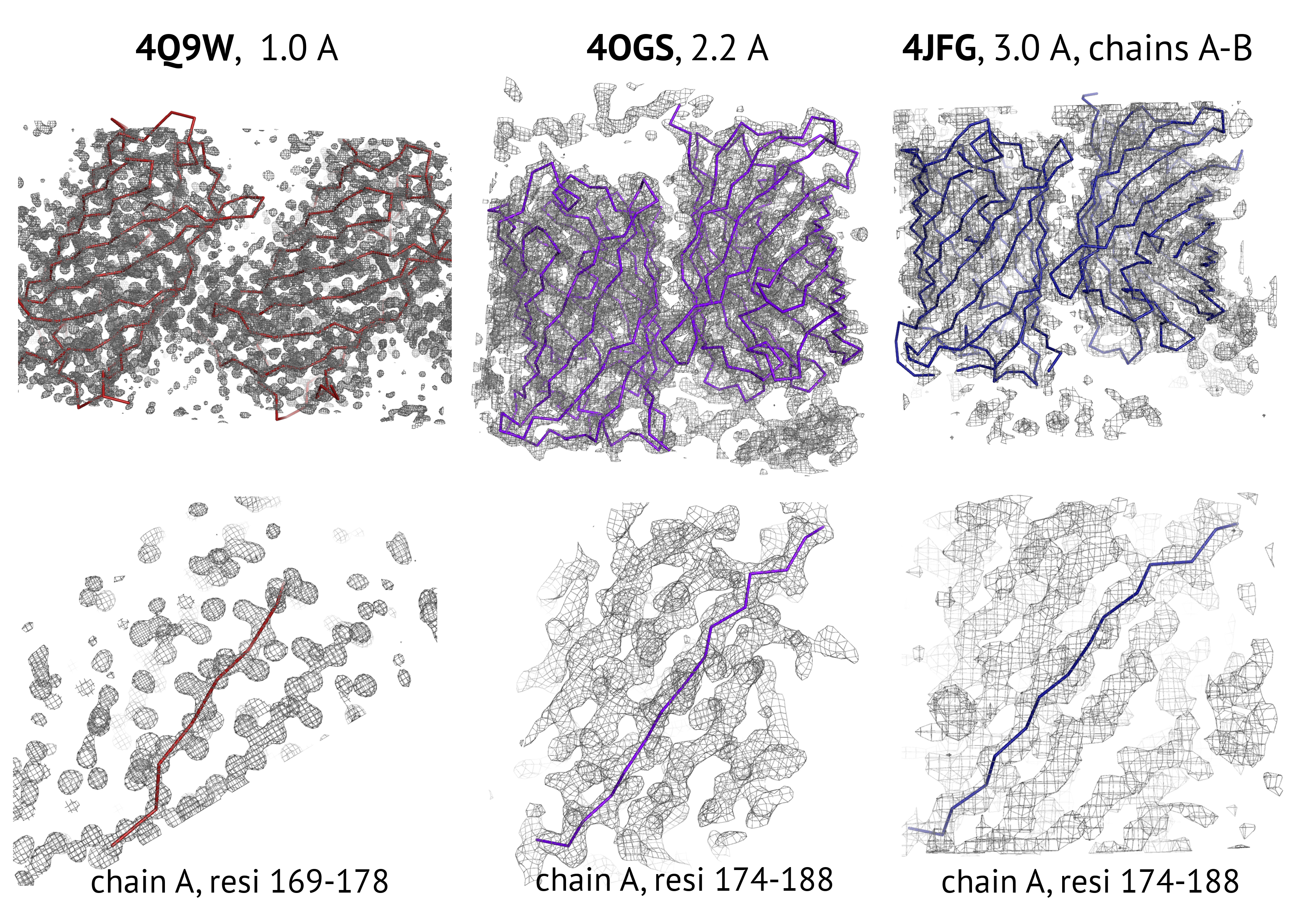
Интересно также рассмотреть электронную плотность этих структур. На любом уровне срезки (проверялись 0.5,1,1.5,2,2.5) заметно, что плотность заметно более сжата для структуры с отличным разрешением. Для структур с плохим и средним разрешением особой разницы не заметно на глаз.
Эти наблюдения неудивительны: температурный фактор как раз отражает степень "размазанности" электронной плотности вокруг атома, чем он меньше, тем более сжатой должна быть плотность.
Выводы
1. Чем лучше разрешение структуры, тем лучше можно различить аминокислоты с альтернативными конформациями. Т.е. в структуре с меньшим разрешением будет больше атомов с коэффициентом заполнения меньше 1.
2. У структуры с лучшим разрешением можно ожидать меньшие среднее и максимальное значение температурных факторов атомов.