Задание 1. Совмещение гомологов по структуре и по последовательности.
Структура для работы: оксидоредуктаза 1ST9. Метод поиска гомологов: PDBeFold. Критерий выбора гомологов: Q-score не более 95, но не менее 70. Количество выбранных гомологов: 7.
Табл. 1 - Выбранные гомологи, характеристики их множественного пространственного выравнивания:
| ## |
Structure |
Nres |
NSSE |
Consensus scores |
| RMSD |
Q-score |
1 |
PDB 1st9:A |
137 |
11 |
0.5557 |
0.9315 |
2 |
PDB 2f9s:B |
136 |
11 |
0.4505 |
0.9492 |
3 |
PDB 2h19:A |
138 |
12 |
0.5220 |
0.9284 |
4 |
PDB 3c71:A |
140 |
12 |
0.5991 |
0.9067 |
5 |
PDB 4nmu:C |
147 |
11 |
0.6624 |
0.8562 |
6 |
PDB 3gl3:A |
141 |
11 |
1.2552 |
0.7967 |
7 |
PDB 3kcm:C |
139 |
11 |
1.4454 |
0.7707 |
|
| Number of aligned residues |
132 |
|
Overall RMSD |
1.322 |
| Number of aligned SSEs |
11 |
Overall Q-score |
0.7297 |
|
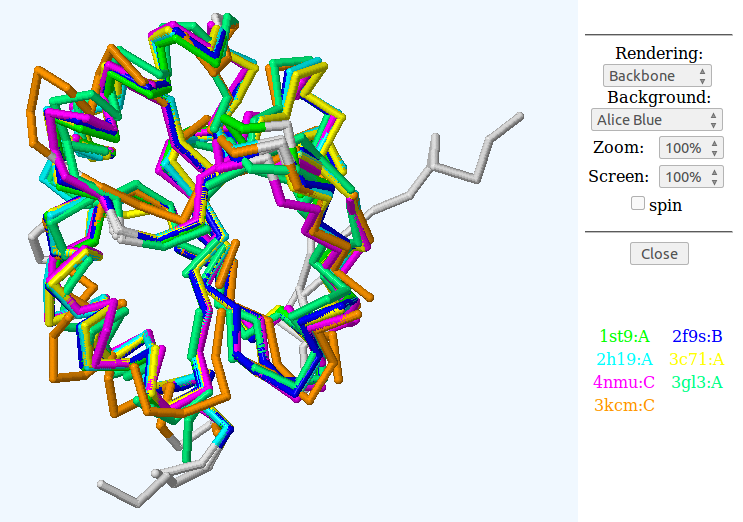
Ссылка на пространственное совмещение (файл с разрешением .jmol): aln.jmol
Ссылка на выравнивание последовательностей по структуре: fasta.seq.
Для сравнения выполнено множественное выравнивание по последовательности с помощью muscle. Ссылка на выравнивание по последовательности: fasta_muscle.seq.
К сожалению, JalView по каким-то причинам не работает в моей версии Linux. Разбираться в странностях ПО - не входит в зачет умений по биоинформатике, поэтому отброшу рекомендацию преподавателей использовать JalView и буду разбираться с выравниваниями вручную.
В целом для требований практикума такой метод вплоне подходит: сразу же обнаруживается множество несоответствий выравнивания по структуре и по последовательности.
Для начала, длины выравниваний несовпадают: 155 для выравнивания muscle и 158 для выравнивания по структуре. Это означает, что количество гэпов заведомо различается.
Действительно, встречаются колонки, где гэп присутствует в выравнивании по структуре, но отсутствует в выраванивании по последовательности. кроме того, в выравнивании по стурктуре можно заметить некоторое количество маленьких букв - т.е. невырованенных позиций.
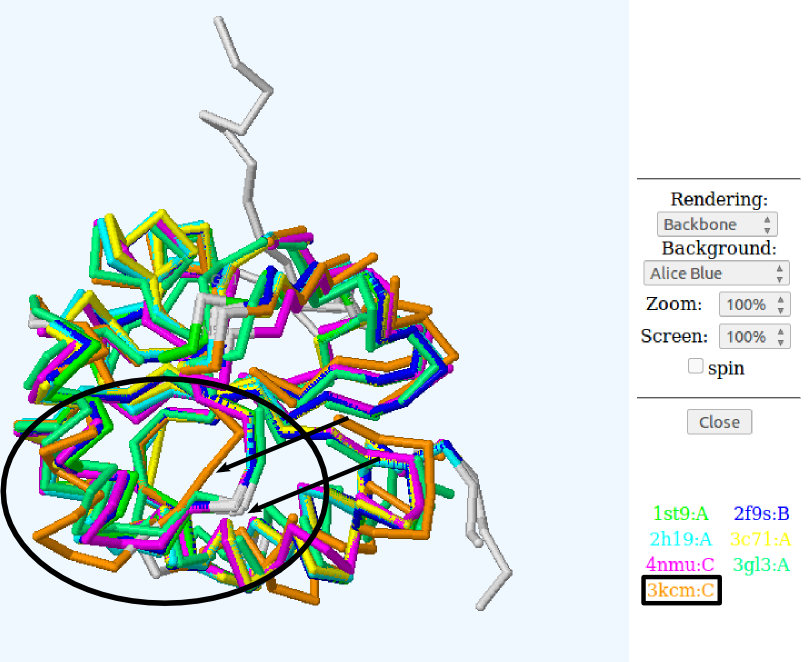
Одно из несоотвтетсвий - позиция 93. В выравнивании по структуре здесь стоит гэп, попоследовательности - непрерывный выровненный фрагмент. Гэп в пространственном выравнивании происходит из-за одной последовательности 3kcm:C (которая, ко всему прочему, наиболее сильно отличается от консенсуса). Этот гэп в колонке компенсируется гэпов в последовательности 3kcm:C через один остаток.
При рассмотрении структуры оказывается, что в этом фрагменте альфа-спираль 3kcm:C лежит немного не так, как соответствующие альфа-спирали других белков. Сделать структурное выравнивание здесь - очень трудно, "неправильные" CA-атомы оказываются сближенными. В данном случае право, скорее всего, выравнивание по последовательности.
Задание 2. Совмещение по заданному выраваниванию.
Задача: сделать совмещение двух структур по выравниванию, заданному через выровненные остатки в бета-тяжах.
Структуры для работы: альфа- и бета- цепи T-антигенного клеточного рецептора. Альфа-цепь представляет собой одиночный бета-лист с неструктурированными петлями по одну сторону от него. (наличие альфа-спиралей не будем учитывать: они менее структурированы, чем бета-тяжи) Бета-цепь представляет собой бета-сендвич.
Для анализа были выбраны: альфа-цепь - 1j8h:d:118-203, бета-цепь - 2bnu:b:114-242.
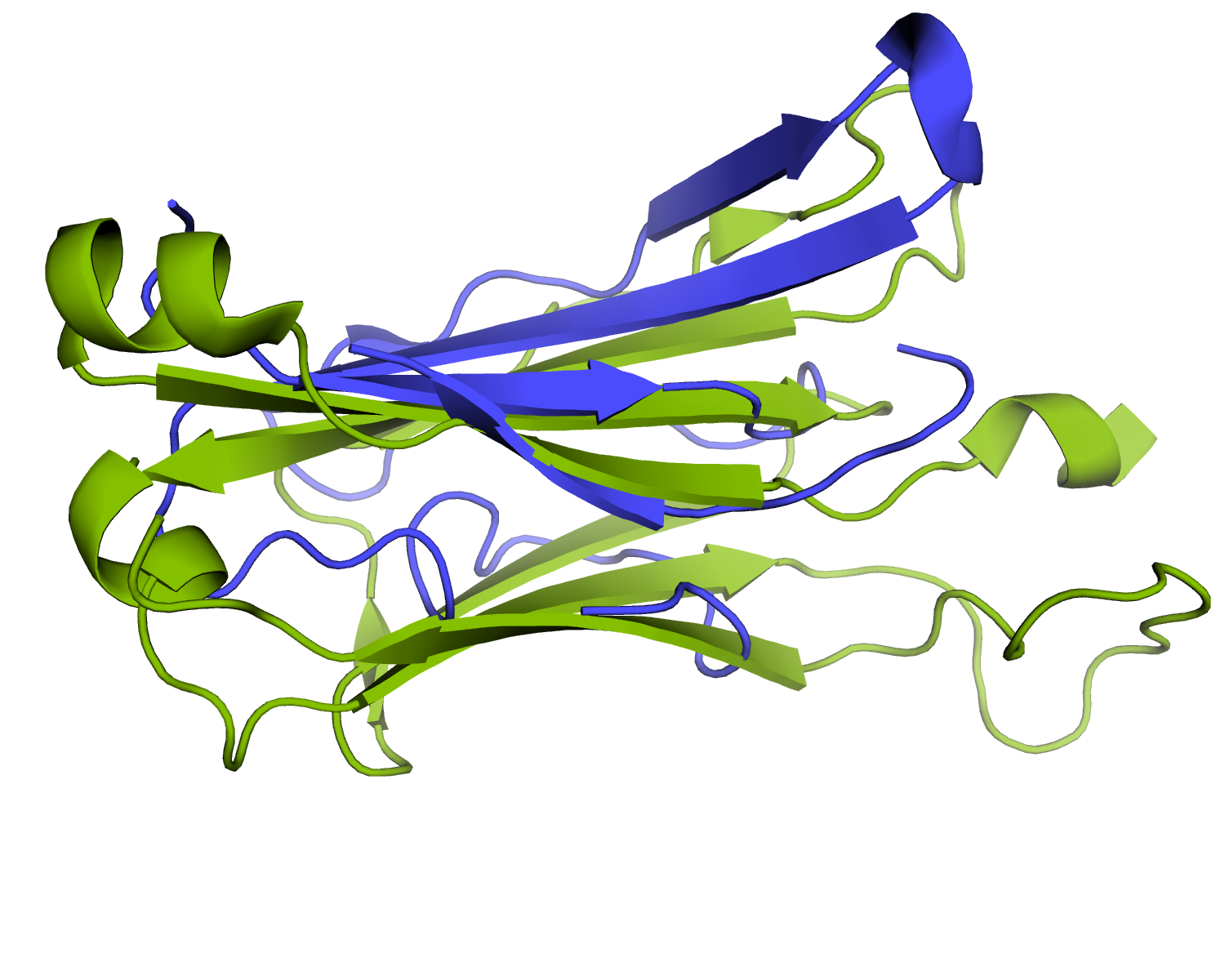
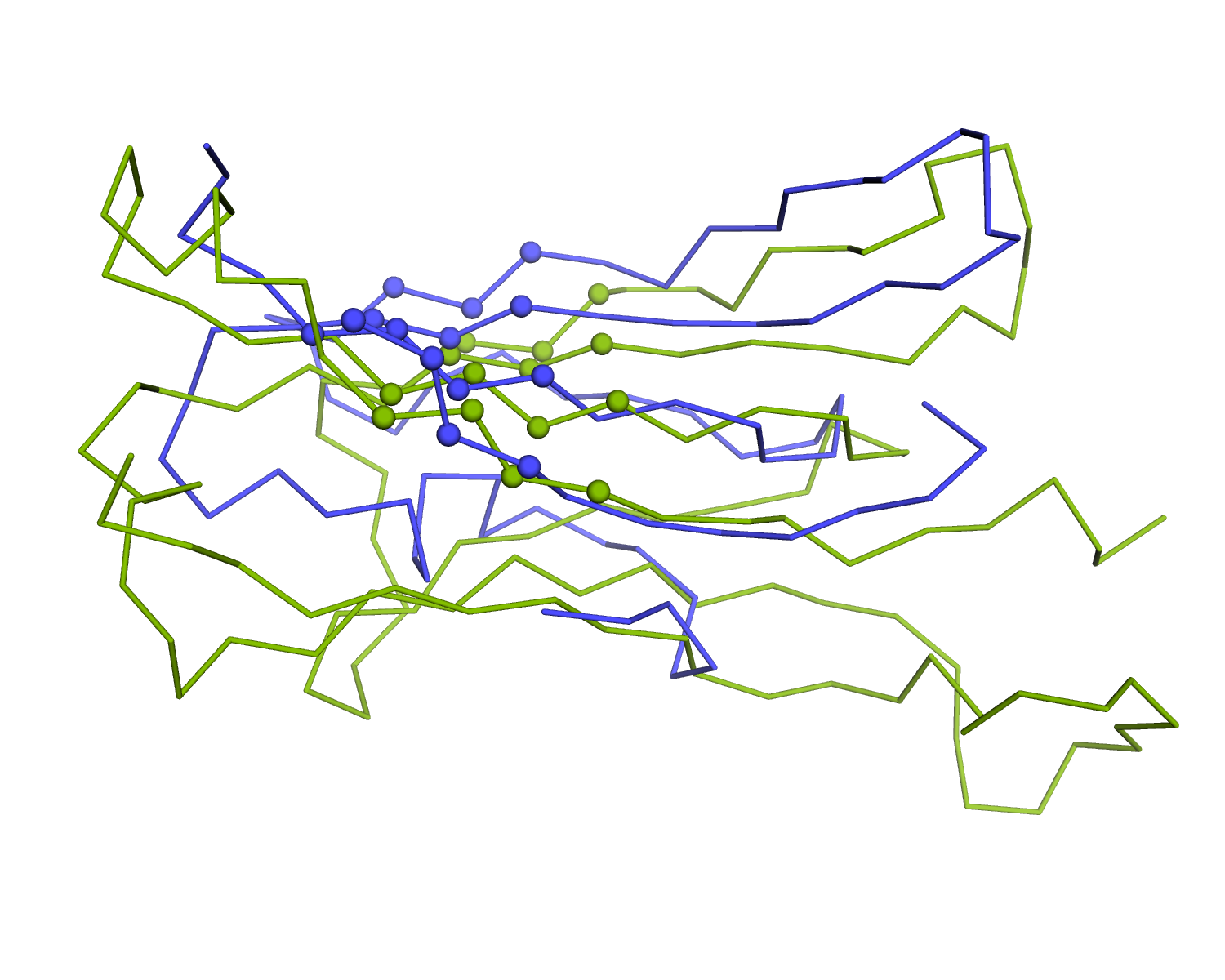
Как показано на рисунке ниже, их прямое выравнивание (методом align в pymol) дает наудовлетворительные результаты. Бета-листы оказываются плохо вырованенными. Неструктурированный фрагмент в альфа-цепи вносит ложную информацию, его не нужно учитывать в выравнивании.
Для выравнивания бета-листов можно использовать SheeP (см. пред. практикум).
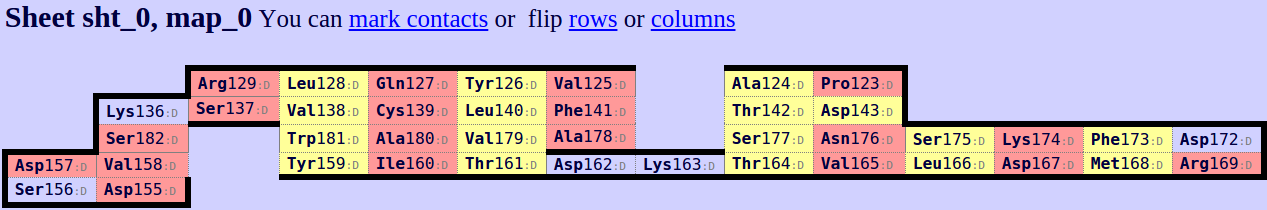
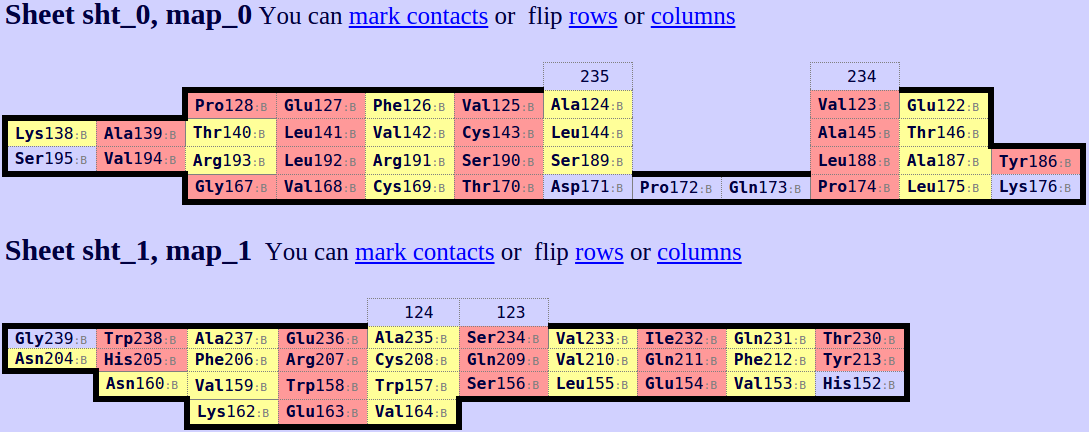
Из карт бета-листов трудно сделать заключение о том, насколько листы из разных структур похожи: требуются дополнительные перестановки.
По крайней мере можно сопоствить количество столбцов в картах (т.е. количество хребтов) и предположить, что карта 0 для бета-цепи (12 хребтов) соответствует карте из альфа-цепи (14 хребтов).
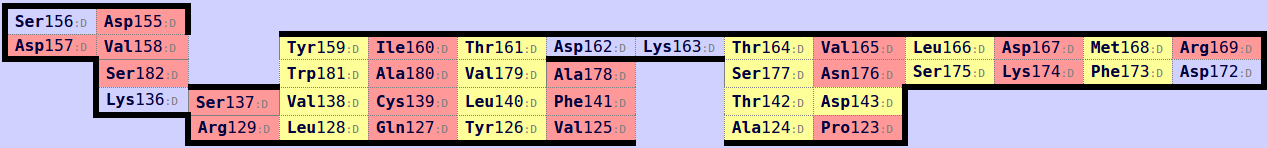
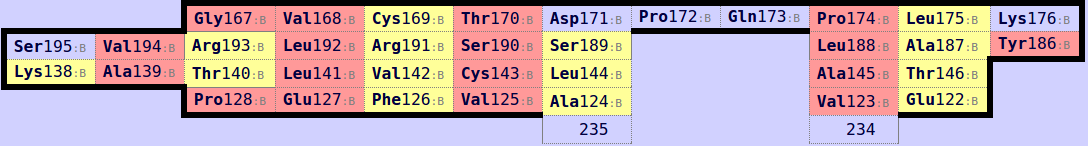
Теперь можно найти консервативный цистеин: 1J8H/CYS`139 (альфа) и 2BNU_mod/CYS`143 (бета). Второй цистеин (169) из структуры бета не подходит, так как расположен на самом краю хребта.
После сопоставления цистеинов получаем однозначное сопоставление остатков двух листов.
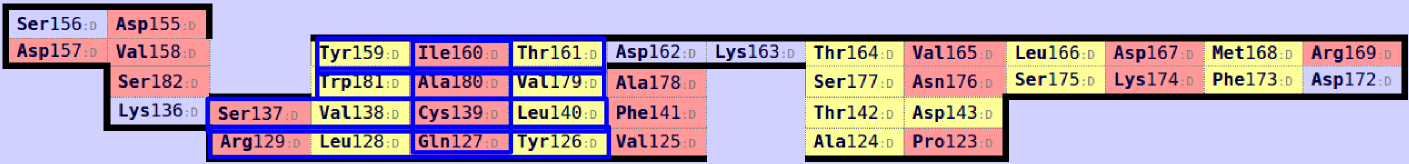
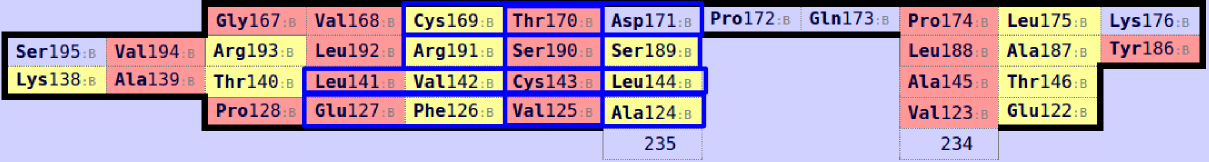
Последовательность команд pymol:
select alpha_align, (resi 137-140 or resi 159-161 or resi 179-181 or resi 126-129) and 1J8H_mod and name CA
select beta_align, (resi 141-144 or resi 189-191 or resi 169-171 or resi 124-127) and 2BNU_mod and name CA
pair_fit alpha_align, beta_align
Важно, что для выравнивания берутся только CA-атомы, хотя можно было бы учитывать и другие атомы остова. Учет радикалов приведет, скорее всего, к различному количеству атомов между выборками. Тогда pair_fit не сможет сделать совмещение, равенство количества атомов в выборках для него - обязательное условие.
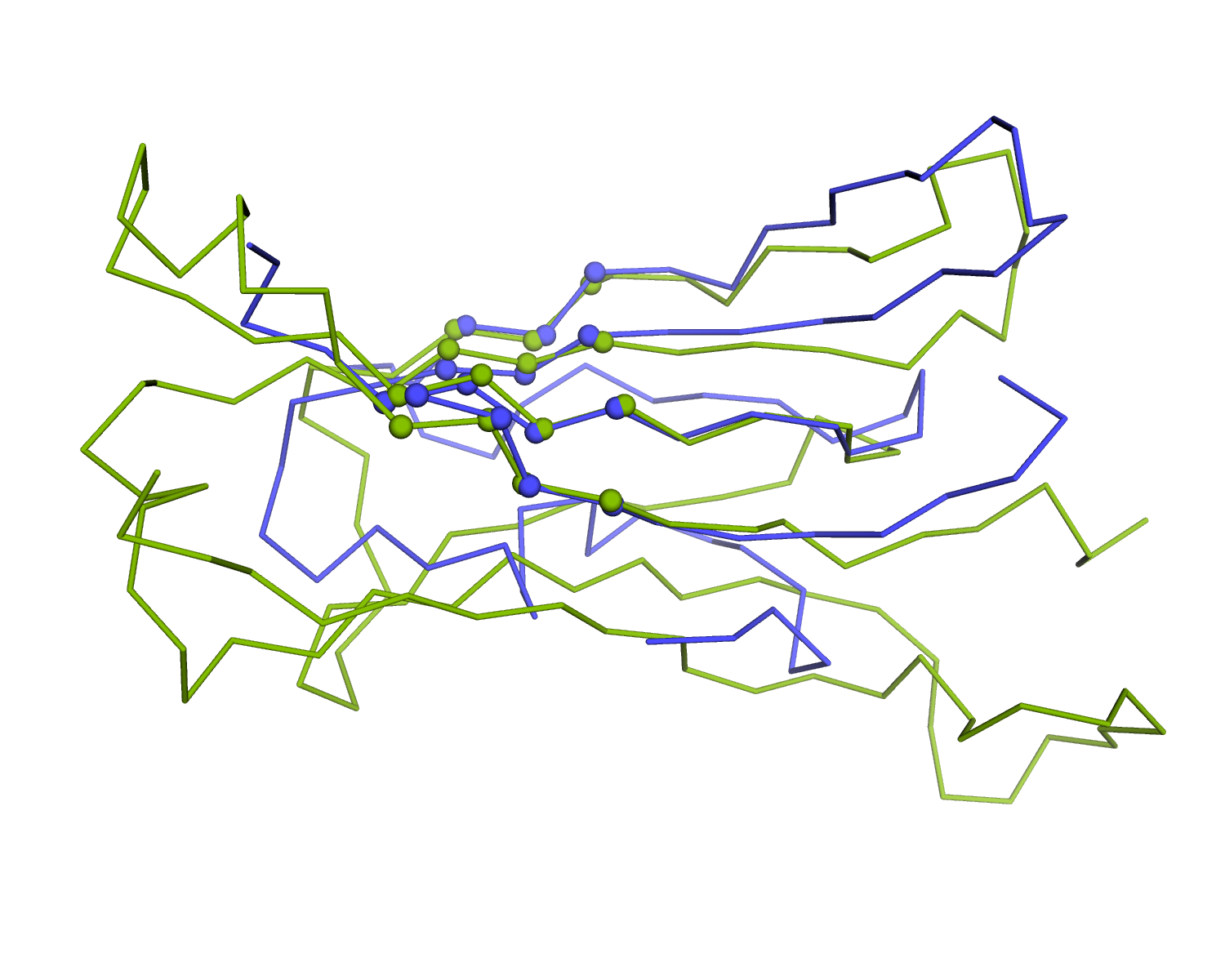
Как видно из рисунков, учет бета-тяжей значительно улучшает выравнивание. Конечно, можно получить выравнивание лучшего качества, если сопоставлять карты полностью, учитывать другие атомы остова и контакты цистеина.
Но даже из такого выравнивания можно сделать заключение о сходстве топологий.
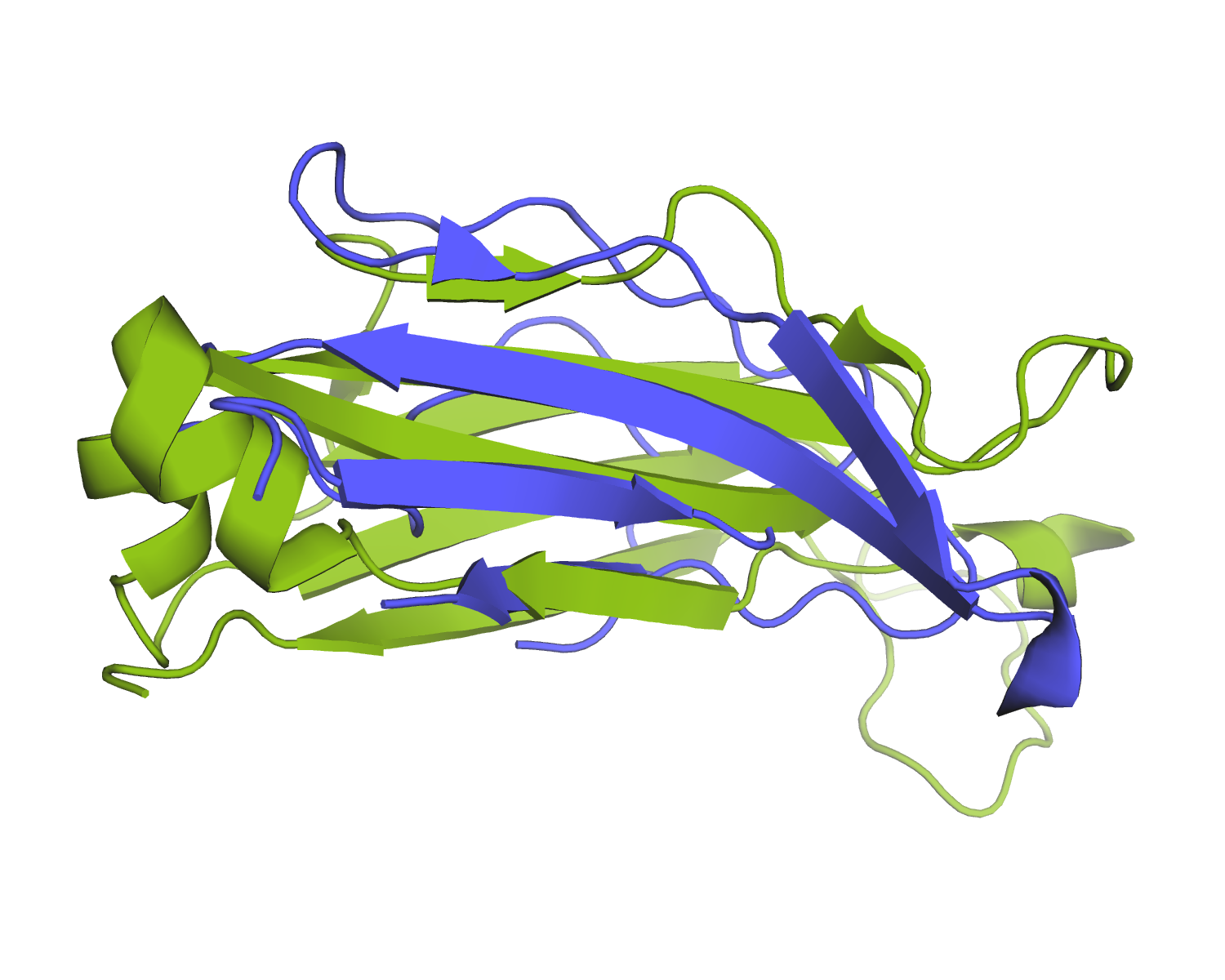
Можно сделать заключение, что топология тяжей сохраняется. Несмотря на то, что длины соответствующих тяжей могут сильно различаться и вместо петель могут быть достаточно протяженные альфа-спирали или неразрешенные участки, закономерности хода цепи по тяжам одинаковы для обоих структур.