Работа с программой Blast
Была дана последовательность белка
Нужно было найти последовательности, гомологичные данной, базе данных Refseq_protein.
Но в этой базе со стандартными параметрами поиска нашлось очень много последовательностей с E-value сильно меньше порога 0.01.
А вот в бызе данных Non-redundant UniProtKB/SwissProt нашлось всего 505 последовательностей (порог E-value - 10)
-Поэтому в дальнейшем я работала с находками по SwissProt
В числе находок бактериальных последовательностей - 407 ; эукариотических - 20 ; архейных - 34.
Для "наилучшей", "средней" и "худшей" последовательности были определены характеристики, представленные в таблице 1.
Таблица1. Сравнение выравниваний по численным хар-кам.
| Белок | Организм | Длина выравнивания | bit score | Процент идентичных колонок | Процент сходных колонок | E-value | |
| Наилучшая | dUTP pyrophosphatase | Mycobacterium avium subsp. paratuberculosis K-10 | 154 | 256 | 95% | 96% | 2e-86 |
| Последовательность из середины списка | dUTP pyrophosphatase | Burkholderia ambifaria MC40-6 | 140 | 69.7 | 37 % | 55 % | 4e-14 |
| Худшая | F-ATPase protein 6 (mitochondrion) | Balaenoptera physalus | 37 | 29.6 | 35 % | 62 % | 8.5 |
360 находок можно условно считать гомологоами исходной последовательности ( Е-value < 0.001 и выравнивания покрывает последовательность на > 70% )
Задание 2
Из полученных находок из задания 1 была выбрана последовательность белка dUTP pyrophosphatase человека с ID: sp|P33316.4|DUT_HUMAN ( с помощью фильтра Formatting options) Так же был осуществлен повторный запуск Blast-а с ограничением на организм (human). Выравнивание не изменилось, однако есть различия в E-value (1e-22 и 6e-24): из-за разного числа последовательностей, с которыми сравнивали исходную (чем больше вариантов, тем более случайна находка и тем больше E-value)
Карта локального сходства Deoxycytidine triphosphate deaminase; Short=dCTP deaminase из Proteus mirabilis HI4320 (gi|238693136|sp|B4ESY9.1|DCD_PROMH) и исходной последовательности
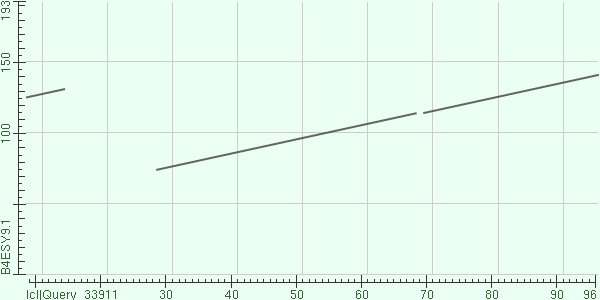
Особенности выравнивания:
- Пробелы соответствуют гэпам
- 2 находки в одном выравнивании (маленький участок в начале query)
- Несмотря на то, что выравнивание кажется "неплохим", e-value = 0.12 говорит об обратном
Задание 4
Через сервер kodomo была создана база данных белковых последовательностей, полученных из множественного выравнивания. Далее был осуществлен поиск гомологов белка
Параметры выравнивания:
- Матрица - BLOSUM62
- Штраф за открытие гэпа - 11
- Штраф за продолжение гэпа - 1
- Max e-value - 11
Параметры предыдущих запросов по Blast-у совпадают за исключением max e-value = 10
Результаты для лучшей находки (ALIAD/1-234):
- Процент идентичных колонок - 26%
- Процент сходных колонок - 38%
- Bit score - 16.2
- E-value - 5.0
Рис2. Лучшее выравнивнаиею
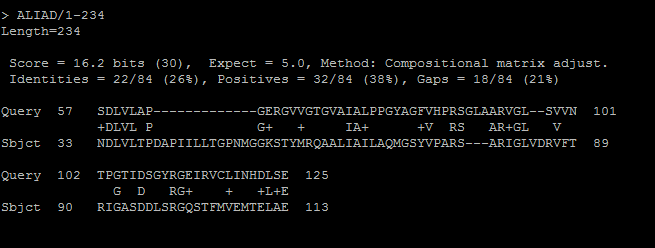
По условному критерию эти последовательности негомологичны
© 2014 Макарова Надежда