Cравнение геномов
Построение карты сходства хромосом двух родственных бактерий
Чтобы сравнить геномы двух родственных организмов, нужно построить парное выравнивание. Результат визуализирован в виде карты генетического сходства. Целью этого задания было найти иллюстрации к некоторым (крупным) эволюционным изменениям в геномах.
Для начала рассмотрим пример выравнивания геномов двух организмов, которые отличаются очень незначительно. Были выбраны организмы Rickettsia typhi str. TH1527
(CP003397.1) и Rickettsia prowazekii str. RpGvF24 (CP003396.1). Это возбудители эпидемического сыпного тифа.
На рис.1 можно увидеть, что в организме Rickettsia typhi близко к origin репликации произошла инверсия, в геноме Rickettsia prowazekii (по вертикали)
произошла вставка.
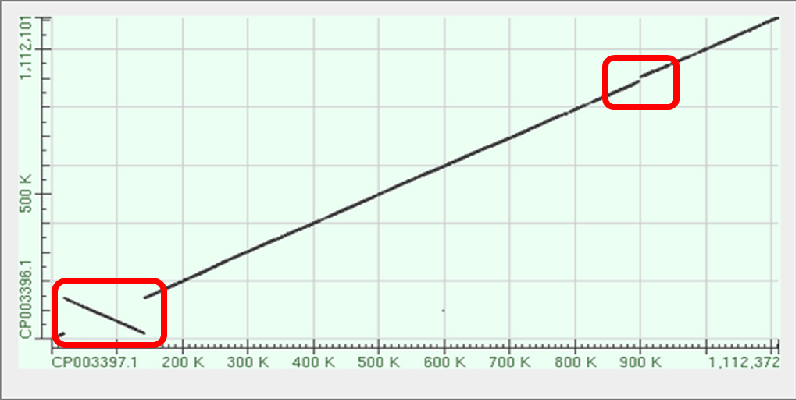
Рис.1 Карта локального сходства двух рикетсий (typhi и prowazekii). Красным выделены иллюстрации крупных эволюционных событий.
Инверсия: примерные координаты (20000-140000). Вставка: примерные координаты (910000-922000).
Теперь рассмотрим более богатое событиями развитие организмов. На рис.2 представлена карта локального сходства Helicobacter pylori 26695 (NC_000915.1)
и Helicobacter pylori J99(NC_000921.1). Здесь одновременно наблюдается инверсия вместе с транслокацией (обозначено красным).
Обнаружено такое явление в 3 местах: 360К-500К; 1.06M-1.16М;1.48М-1.5М. Заметим, что данная карта сообщает, что 2 больших участка (большие красные прямоугльники)
в геноме поменялись местами. В фиолетовом квадрате можно различить вставку в Helicobacter pylori J99 и инверсию участка генома Helicobacter pylori 26695
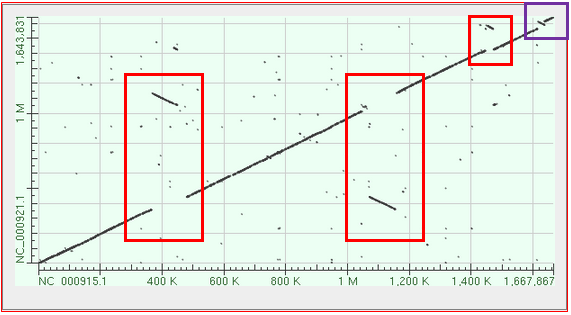
Рис.2 Карта локального сходства Helicobacter pylori 26695 и Helicobacter pylori J99.
Описание сходства и различий геномов близкородственных бактерий с помощью NPG
Программа NPG-Explorer строит пангеном - множественное выравнивание геномов близкородственных бактерий и архей в специальном формате. Особенность - разделение выравнивания на блоки в зависимости от встречаемости и длины фрагмента.
Для сравнения были выбраны следующие штаммы Rickettsia rickettsii В скобочках индентификаторы RefSeq
- Rickettsia rickettsii str. Brazil CP003305.1
- Rickettsia rickettsii str. Morgan CP006010.1
- Rickettsia rickettsii str. Iowa CP000766.1
- Rickettsia rickettsii str. Arizona CP003307.1
Далее был создан файл с информацией, откуда брать геномы и аннотации генов. В параметрах npge изменила Min_Identity на рекомендованные '0.89'; Workers = 1.
В результате работы NPG explorer было получено много файлов с аналитической информацией о пангеноме, который был визуализирован c помощью программы gnpge.
Анализ полученных данных
Глобальные блоки
Это блоки состоят из последовательно идущих во всех геномах s-блоков, перемежающихся блоками других типов (r-, h-, u- и m-)
Всего найдено 5 глобальных блока. Их выравнивание представлено на рис.3.

Рис.3. Выравнивание g-блоков
В дальнейшем геномы будут названы в зависимости от штамма. Из данного выравнивания видно, что геномы Arizona, Iowa и Morgan имеют одинаковую последовательность g-блоков, между которыми находятся одинаковые i-блоки. В то время как Brazil взят в обратном направлении ("-" в названии генома в выравнивании). Это означает, что геном Brazil был секвенирован по комплементарной цепи. Еще нужно заметить, что хотя последовательность и комплементарна, но порядок блоков g-блоков в ней не соответсвует таковому, но перевернотому у трех остальных. Это значит, что последовательность кольцевой хромосомы Brazil начинается не стого места, что у остальных трех. Это предположение подтверждается, если взглянуть на карту локального сходства Brazil и одного из трех других геномов (в данном случае c Arizona) - см.рис.4
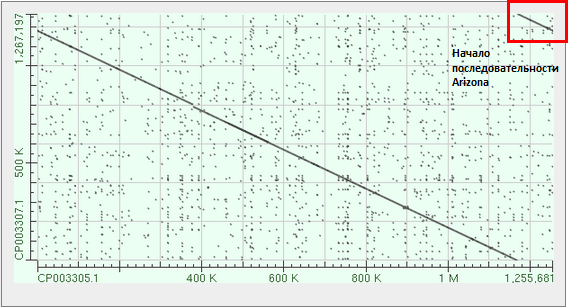
Рис.4 Карта локального сходства геномов Brazil и Arizona.
Стабильные блоки
Такие блоки образуются из фрагмента, который есть в единственном количестве у каждого из геномов
Количество - 108; суммарная длина в среднем - 1240793; процент от длины генома в среднем - 98.43%;
процент консервативных позиций в объединенном выравнивании s-блоков - 99.8923%
Это означает очень высокое сходство между геномами.
Блоки с повторами хотя бы в одном геноме.
Стоит заметить, что повтор фрагмента еще не означает повтор гена
Пример: r8x126. Содержит 8 фрагментов: Arizona - 2, Morgan - 2, Brazil - 2, Iowa - 2. Имеет длину 126. Процент консервативных колонок - 96.82. Содержит участки 5 генов:
- CDS RrIowa_0849_RrIowa_0849 tetratricopeptide repeat family protein (Iowa), 117 bp <
- CDS RPO_04500_RPO_04500 replicative DNA helicase (Arizona), 306 bp <
- CDS RPN_02470_RPN_02470 aromatic acid decarboxylase (Brazil), 306 bp <
- CDS RrIowa_0952_RrIowa_0952 replicative DNA helicase (Iowa), 273 bp <
- CDS RRM_04265_RRM_04265 replicative DNA helicase (morgan), 306 bp <

Рис.5 Блок r8x126. Белым выделены нуклеотиды, входящие в ген.
Мне показалось странным, что у Iowa в втором фрагменте программа аннотировала пептид, в то время как у идентичных фрагментов из Arizona и Morgan ничего не было выявлено. Во-первых, стоит понять, что это за ген. Это ген пептида, образующего повторяющийся мотив, который служит связующим звеном в комплексе белков. В записях GenBank этот ген не аннотирован (просто назван участок - repeat region). Генное окружение одинаково у всех трех геномов на этом участке и на этой цепи. Не известно, почему программа аннотировала только Iowa этот пептид.
Полустабильные блоки
Выбран блок h3x9668. В нем присутствут 3 фрагмента: Iowa, Morgan, Arizona. Длина 9668. Крупная делеция в Brazil. На нем содержится 13 генов.
Выбран блок h3x857. В нем содержатся фрагменты Iowa, Morgan, Arizona. Длина 857. 9 генов.
Блок h3x121n1 представлен на рис.5
Уникальные блоки
Единственный блок - Brazil u1x102. Генов не представлено.
Прогнав по BLAST, были найдены находки из Arizona Iowa и Morgan, только по перевернутой псоледовательности и с заменой 1 нуклеотида.
Если посмотреть на выравнивание блоков в районе этого блока (см.рис.6) и на последовательность фрагмента внутри блоков,
можно заметить, что они различаются направлением и несколькими нуклеотидами. Здесь уникальный блок не отражает вставки или другого изменения
(судя по карте локального сходства).

Поиск расхождений между аннотациями генов из одного блока
- 1. В трех геномах ген аннотирован (Morgan, Arizona, Brazil), а в оставшемся - не признается за ген.

Начало участка, в котором не аннотирован ген у Iowa. У Arizona и Morgan там гипотетические белки. У Brazil - ген устойчивости.
- 2. В геномах Morgan Iowa и Arizona на данном участке закодированы рибосомальные белки. А в Brazil некий активный фермент.
Также гены рибосомальных генов различаютcя по длине, а, следовательно и по началу в геноме. Все гены на прямой цепи.

По рассматриваемым примерам становится ясно, что наиболее схожи геномы Arizona и Morgan (по расхождениям в аннотациях и положениях генов). А Iowa и Brazil выбиваются, но не очень схожи друг с другом. Главное, что нужно понимать, что геном Brazil представлен комплементарной последовательностью. Из-за этого функция белков, закодированных в казалось бы ортологичных генах, сильно различается.
Надо сказать, что я все-таки еще проверяла себя по картам локального сходства, работая со множественным выравниванием в виде блоков. Однако это делает возможным быстро находить важные участки, характеризующие определенный геном.
В заключении, я хотела бы заметить, что сначала было немного сложно связать последовательность блоков с эволюционными событиями. Здесь было представлено сравнение очень близких геномов, отличия которых очевидны. При сравнении более разнородных геномов в визуализаторе было сложно что-то однозначно определить (по-началу). Например, чтобы найти интересный случай, связанный с горизонтальным переносом, которого мне не встретилось, я выбрала четыре разных видов рикетсий. И в том пангеноме было много уникальных блоков, но практически все они были, во-первых, из одного генома, во-вторых, состояли из фрагментов, непопавших по какой-то причине в s-блоки ( по Blast они были во всех 4 геномах). Поэтому программа лучше подходит для сравнения близкородственных организмов.
© 2014 Макарова Надежда