Работа с программами EMBOSS
Упражнения
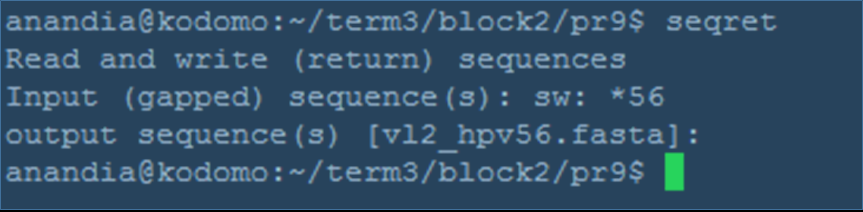
- seqret - объединяет несколько fasta-файлов в один. Здесь: был получен fasta-файл содержащий псоледовательности белков, чей идентификатор в базе
SwissProt (проверенных белков) заканчивается на 56.
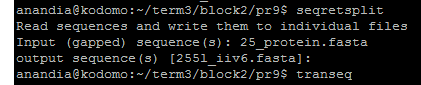
- seqretsplit - разбивает один файл на несколько fasta-файлов. Здесь: из файла, содержащего последователньости белков,
чей идентификатор начинается с 25
было получено 5 fasta-файлов, содержащих по одной йпоследовательности.
- transeq - транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные, используя указанную таблицу генетического кода.
Здесь: последовательности в файле ex3.fasta Транслирует в один файл согласно бактериальной таблице генетического кода.

- transeq - транслировать кодирующие последовательности, лежащие в одном fasta файле, в аминокислотные в 6 рамках. Здесь: последовательности в файле ex5.fasta транслируется в один файл в 6 рамках.
- seqret Программа seqret часто используется для перевода выравнивания из одного формата в другой. Здесь: из выравнивания в fasta-формате получили
выравнивание формата aln.

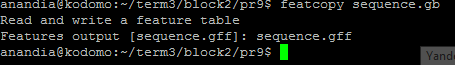
- featcopy Переводит аннотации особенностей в записи формата .gb в табличный формат .gff
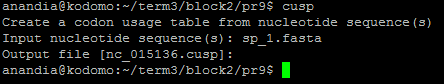
- cusp Определяет частоты кодонов в данных кодирующих последовательностях. Результат
здесь.
- compseq Определяет частоты динуклеотидов в данной нуклеотидной последовательности и сравнивает их с ожидаемыми. Результат
здесь.

Сравнение аннотации генов белков в одной хромосоме бактерии с трансляциями длинных открытых рамок считывания.
Введение
Открытая рамка считывания (Open Reading Frame - ORF) - это последовательность нуклеотидов, разбитая на триплеты для трансляции в последовательность аминокислот. Начинается инициирующим кодоном, заканчивается терминирующим. В зависимости от стартовой точки на двух цепях существует 6 рамок считывания. Если внутри рамки считывания встречается стоп-кодон, то белок с нее не считывается (рамка блокирована). В данном задании нужно получить все возможные трансляции открытых рамок, отвечающих заданным параметрам, и сравнить с аннотациями генов. Аннотации генов - это нахождение или предсказание функционально важных участков генома (например, кодирующих РНК, белок или регуляторный участок). Они создаются путем сравнения участков генома либо с транскриптомом, либо с протеомом. Иногда функцию участка генома становится вазможно узнать, выравнивая последовательность с уже известными геномами. Есть и другие подходы. В данной работе можно наглядно увидеть, что наличие открытой рамки (найденной программой) считывания еще не гарантирует существование белка.
В качестве объекта изучения была выбрана бактерия, изучаемая в первом семестре, Amycolatopsis orientalis HCCB10007
Трансляция открытых рамок считывания
C сайта NCBI я скачала файл в формате GenBank (gb) с аннотированной последовательностью кольцевой хромосомы ДНК (ACCESSION: NC_021252).
Далее, использовав программу getorf пакета EMBOSS, я получила список трансляций открытых рамок считывания данной хромосомы.
Описание команды: getorf -sequence sequence.gb -outseq translation.txt -table 11 -minsize 180 -circular Y -find 0
.
Что означает: данная программа будет искать в файле (-sequence sequence.gb) с последовательностью нуклеотидов кольцевой хромосомы (-circular Y)
рамки считывания от стоп-кодона до стоп-кодона (-find 0) длиной не менее 180 нуклеотидов (-minsize 180) и транслировать их в соответствии с бактериальной таблицей генетического кода (-table 11).
Выходной файл представлен здесь.
Затем, чтобы сделать полученные данные более доступными, была использована программа infoseq пакета EMBOSS.
Описание команды: infoseq translation.txt -outfile table_translation.txt -only -name -length -description
. Что означет: данная программа,
проанализировав файл (translation.txt), запишет в файл (-outfile table_translation.txt) только (-only)
информацию об ID открытой рамки (-name), координатах в геноме (-description) и длине трансляции в остатках (-length).
Выходной файл представлен здесь.
C помощью следующего скрипта полученные данные были отформатированы. В итоге, былв создана таблица трансляций рамок считывания.
Получения списка аннотированных генов белков
В браузере Genome NCBI была найдена и скачана таблица аннотированных генов белков. C помощью скрипта полученные данные были отформатированы. В итоге, былв создана таблица аннотированных генов белков . Так же был скачан файл c последовательностями всех белков, закодированных в хромосомной ДНК Amycolatopsis orientalis HCCB10007
Изучение полученных данных
Отсортированная по очередности в последовательности и по принадлежности к прямой или обратной цепи таблица представлена здесь. Ярко-желтым представлены рамки считывания, найденные программой getorf в прямой цепи. Бледно-желтым - в обратной. Ярко-синим показаны предсказанные белок-кодирующие гены в прямой цепи, бледно-синим - в обратной.
Далее приведены несколько примеров расхождения между трансляциями открытых рамок, выполненных программой getorf и аннотированными участками генома, а так же их объяснение
- 1. Для иллюстрации стандарта было решено пойти по последовательности и найти наименее расходящиеся участки. Выбран следующий фрагмент - см.рис1.
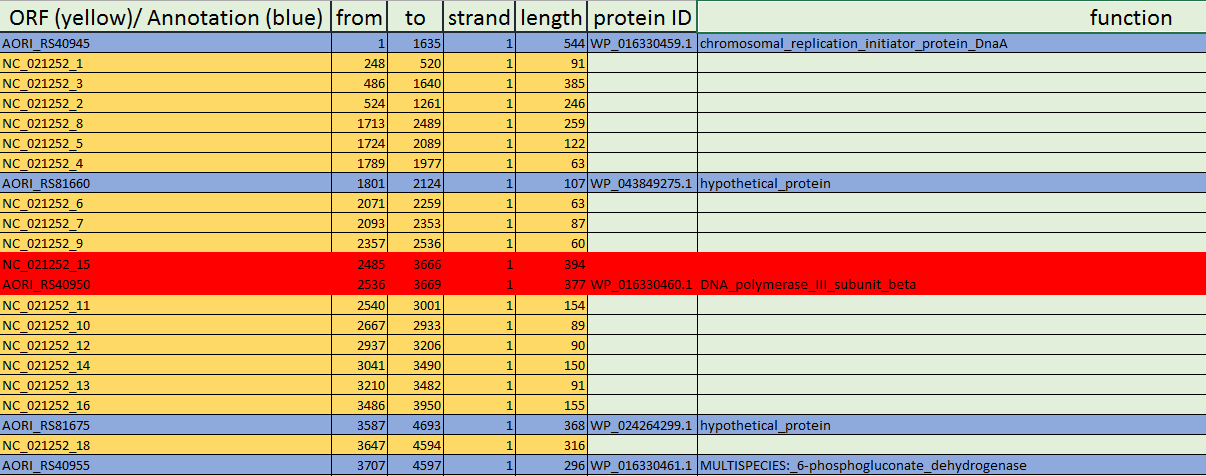
Рис.1 Первое выбранное расхождение. Выделено красным.
Расхождение в конце последовательности всего в три нуклеотида! ORF длиннее аннотированной последовательности (алгоритм getorf ищет от стоп-кодона до стоп-кодона, а в аннотированной - от старт-кодона до стоп-кодона => меньше). Взглянув на последовательность (см.рис2), стало понятно, что различие в три нуклеотида наблюдается из-за того, что аннотированная запись включила в координаты стоп_кодон TGA. Данное расхождение наиболее часто встречается и служит критерием достоверности правильного выбора ORF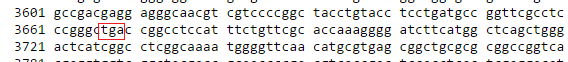
Рис.2 Расположение стоп кодона 3666-3669. Выделено красной рамкой. - 2. Во втором примере представлена проблема, связанная с длинной ORF. Из рис.3 можно заметить, что наиболее подоходящая
последовательность ORF1 прерывается стоп-кодоном. Следом за ней идет довольно большая ORF2. В связи с этим возникает несколько вопросов: почему в предсказанном гене
hypothetical protein, "игнорируется" стоп-кодон, которым заканчивается ORF1 и почему в большой ORF2 "игнорируется" стоп-кодон,
которым заканчивается аннотированная последовательность. Это объясняется тем, что getorf ищет те рамки, размер которых больше указанного (180 н).
Поэтому он "разбивал" последовательность, избегая коротких ORF. Пусть в последовательности, кодирующей hypothetical protein есть последовательность TGA,
но так как там другая рамка считывания, то она не блокируется. Также и в ORF2.
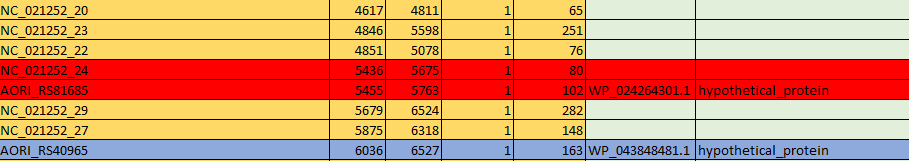
Рис.3 Второе выбранное расхождение. Выделено красным.
- 3. Еще один пример расхождения данных из-за длины последовательности. На этот раз аннотированной. Так как длина белка меньше 60, программа не нашла
хотя бы немного похожей ORF.
Рис.4 Третье выбранное расхождение. - 4. В комплементарной цепи ДНК так же есть большое количество расхождений 1-ого типа. Только теперь первые позиции отличаются на 3 нуклеотида (см.рис.5)
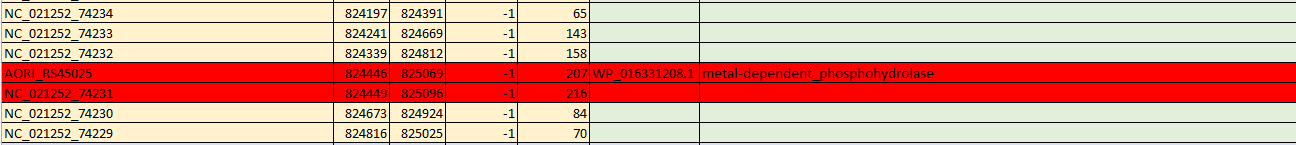
Рис.5 Четвертое выбранное расхождение. Выделено красным. - 5. Этот пример интересен тем, что концы двух ORF (с номерами 20597 и 20599) правильно определены. Однако одна ORF(20597)
включает целых две кодирующих последовательностей. Аннотированные участки идут непрерывно друг за другом. getorf определил правильно stop-кодон первого, но почему,
определив правильно stop-кодон второго ( => при этом рамка считывания не поменялась: так как аннотированные последовательности образуют тандем) программа не разделила две ORF? Вероятно, это ошибка программмы.

Рис.6 Пятое выбранное расхождение. Выделено красным.
Другие схожие примеры расхождений представлены в Excel-файле. Они выделены красным.
Перекрывание антипараллельных рамок считывания
Чтобы найти примеры данного явления, нужно понимать, что getorf использует для обратной цепи нумерацию прямой, но читает ее в обратном порядке!. Будем считать перекрывание "нужным", если его длина больше 150н. Я решила выбрать те антипаралельные ORF, которые содержат аннотированные последовательности и перекрываются , так как рассмотрение любых пересечений антипараллельных рамок не имеет смысла, так как программа ищет все возможные рамки, но не факт, что они реализуются в геноме. Для этого я выбрала два рядом лежащих, но на противоположных цепях довольно длинных белка (на других не наблюдалось). Данные рамки изображены на рисунке7. Схему перекрывания можно посмотреть на рисунке8.
Рамка с номером 77511 на обратной цепи имеет длину 483 и содержит последоватленьость, кодурющию белок malyl-CoA_lyase (закрашенный зеленый прямоугольник) перекрывается с рамкой с номером 300 имеющейц длину 766 и кодирующей белок serine/threonine_protein_kinase(закрашенный серый прямоугольник) 355 нуклеотидами. Показалось интересным, что для каждой белок-кодирующей последовательности есть рамка считывания, но на противоположной цепи (выделены желтым). Причина такого перекрывания - алгоритм getorf(стоп-стоп). При распозновании старт кодона данного перекрытия не было бы.
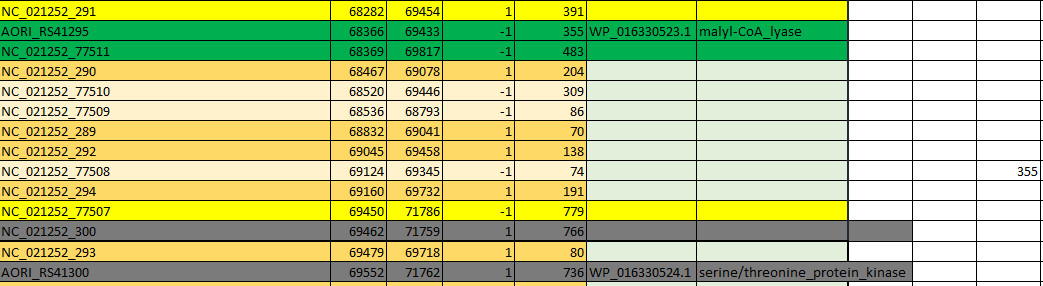
Рис.7 Пример перекрывания антипараллельных рамок считывания
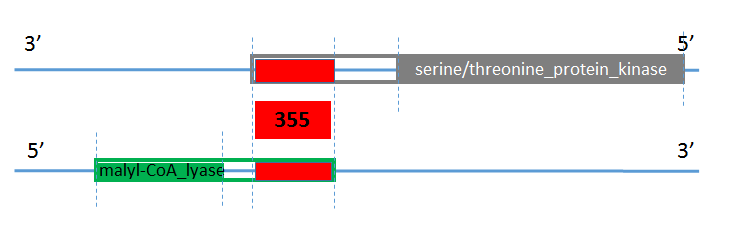
Рис.8 Схема перекрывания антипараллельных рамок считывания
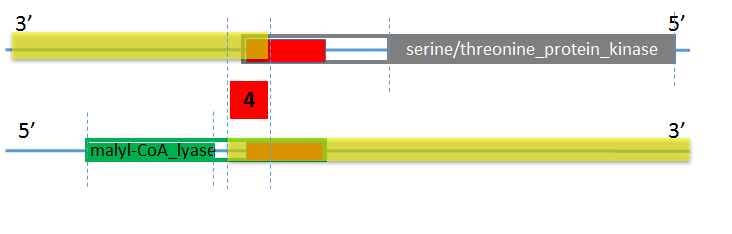
Рис.9 Схема перекрывания антипараллельных рамок считывания.

Рис.10 Последовательность участка, который присутствует антипараллельных ORF, но не включается ни в одну аннотированную последовтельность.
Чтобы найти другой пример перекрывания, когда рамка считывания перекрывается вследствии перекрывания генов, нужно найти перекрывания аннотированных последовательностей. Выбрав перекрывание соседних антипаралельных аннотированных последовательностей (с помощью Excel), я выяснила что больше всего они перекрываются в самом начале. Но на основе этих данных мне не удалось найти подходящий пример перекрывания ORF: везде где аннотированные последовательности перекрываются более чем на 150н, там рамка определена неподходяще (т.е. разбита с целью избежать коротких ORf). Однако нужно сказать, что такое явление довольно роспространено у организмов с маленькими геномами (бактерий и вирусов). Они вынуждены плотно упаковывать свои гены => происходит перекрывание.
© 2014 Макарова Надежда