Ресеквенирование
У нас есть рефересная последовательность 11-ой хромосомы человека. Есть риды, полученные при секвенировании 11 хромосомы но у другого человека. Задача: найти однонуклеотидные изменения (делеция, вставка, замена) - полиморфизмы в вновь секвенированной хромосоме человека.
Подготовка чтений
Для этого нужно подготовить чтения. Убрать те, качество которых нас не устраивает. Чтобы наглядно увидеть изменения получим графическое изображение (Рис.1),
которое покажет понуклеотидное качество секвенирования.
fastqc chr11.fastq
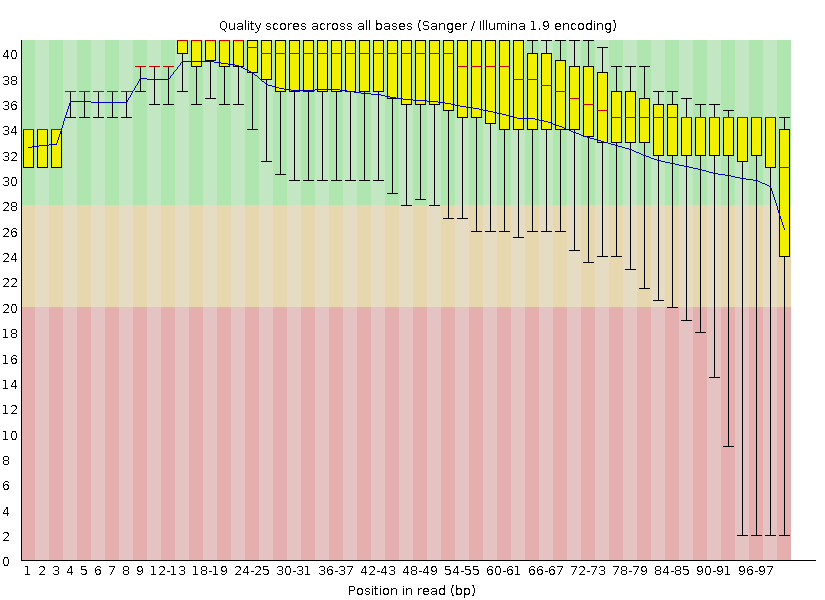
Рисунок 1 Графическое изображение понуклеотидного качества секвенирования до фильтрации по качеству и длине
Далее очистим наши риды с краев и оставим те, качество которых больше 20, а длина не меньше 50. В этом поможет Trimmomatic.
java -jar /usr/share/java/trimmomatic.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:50
Что же изменилось? Во-первых у нас удалилось 134
последовательности: было 4198, стало - 4064ю Во- вторых это мы сможем узнать опять при помощи контроля FastQC (Рис. 2).
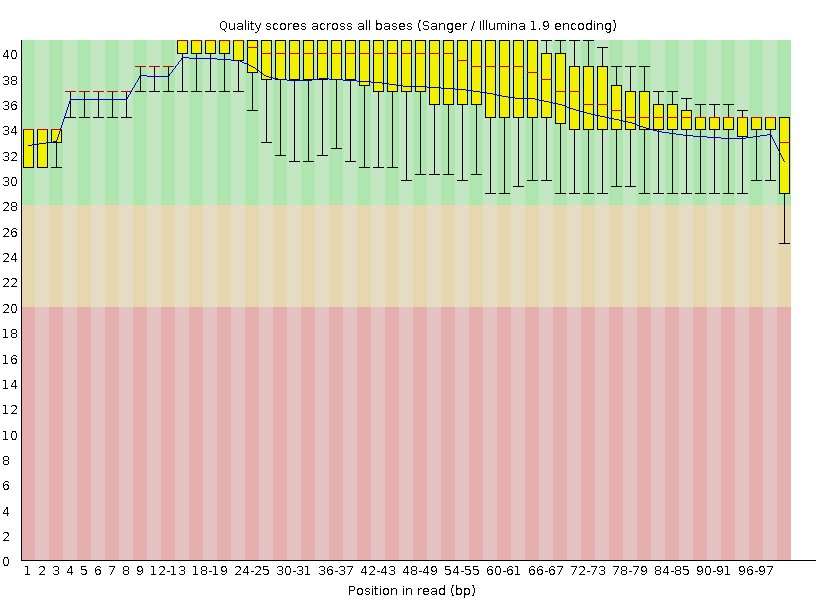
Рисунок 2 Графическое изображение понуклеотидного качества секвенирования после фильтрации по качеству и длине
Нужно пояснить, что у нас все риды длиной 100 - это параметр секвенатора. За счет их большого количества машина может относительно быстро секвенировать целый геном.
На этих изображениях показана информация о распределении качества прочтения одного нуклеотида (вертикальная ось - положение этого нуклеотида в риде: от 1ого до 100).
Качество нуклеотида - это величина, которая обратно зависит от вероятности ошибки. Распределение понимается так:
в первом нуклеотиде каждого рида можно сделать ошибку секвенирования с некоторой вероятностью. Ридов очень много.
Соответственно появятся значения качества прочтения (т.е. вероятности сделать ошибку), которые чаще встреяаются или почти невстречаются.
Это информация и отражается в каждом столбце картинки. В хвостах (тонких черных полос) мы можем найти значения качества, которые редко встречаются,
в желтой области мы имеем распределение - "горку", на вершине которой мода - то значение, которое встречается чаще всего.
Красным обозначено среднее значение.
На основании этой информации мы теперь можем понять что изменилось.
Во-первых, укоротились хвосты с маленьким значениеи качества секвенирования. Желтая область тоже поуменьшилась.
Теперь у нас нет никаких ридов в красной области (собственно, что мы и хотели).
Также в выдаче находятся и другие изображения.
| До обработки trimmomatic | После обработки trimmomatic | Различия |
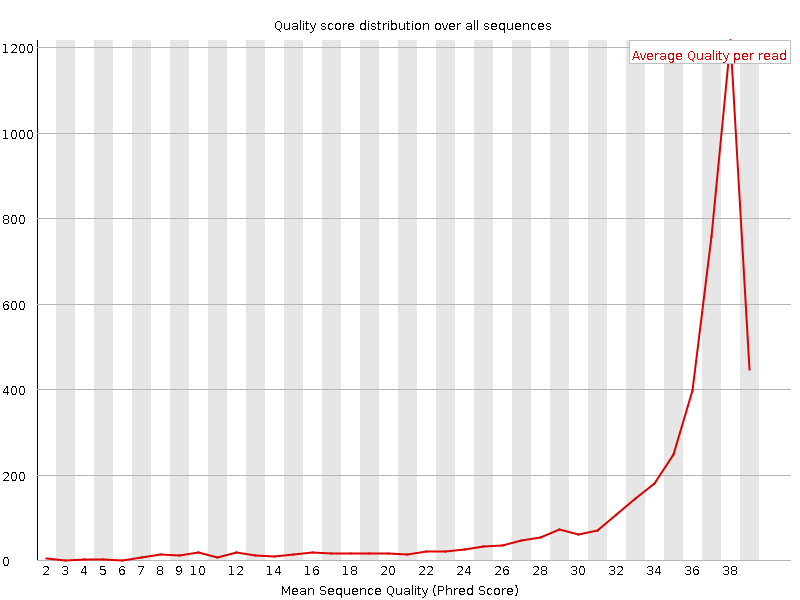 |
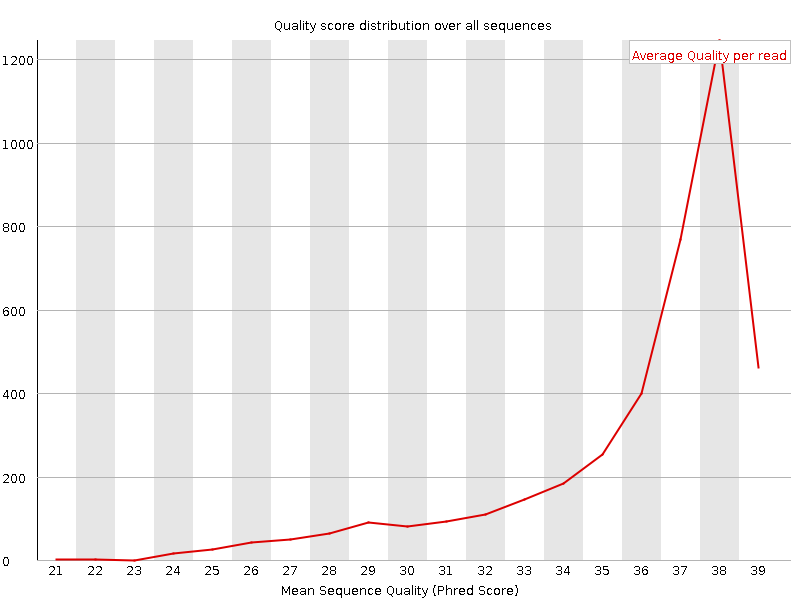 |
Картинка отражает распределение ридов по качеству. Хвост в районе 20 - 30 (по оси качества) заметно изменился. Их количество (ридов с низким качеством) свелось к минимальному после обработки. |
 |
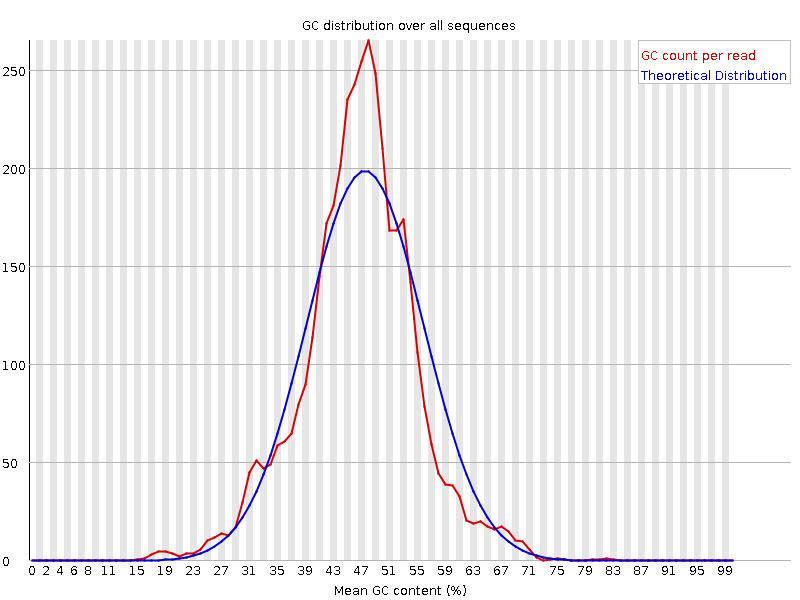 |
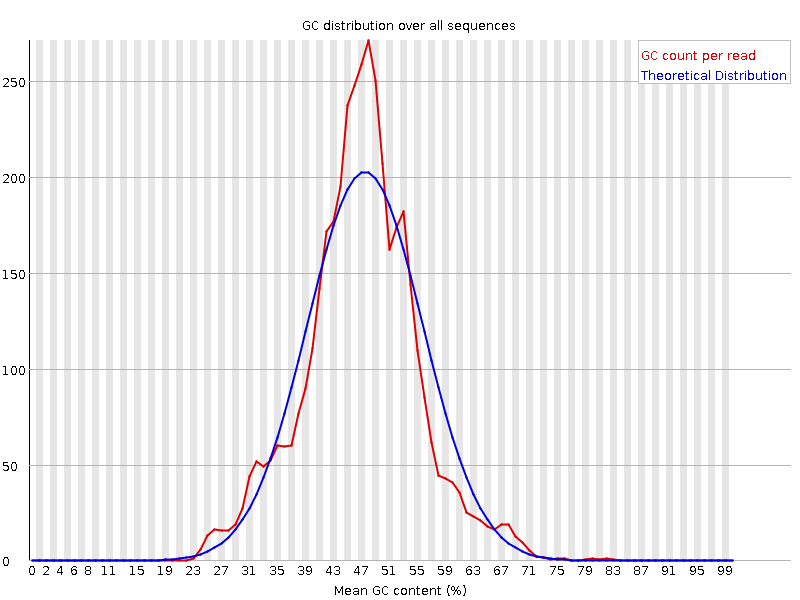
Картинка отражает распределение содержания GC (т.е. процентное содержание нуклеотидов гуанина или цитозина - т.к. они образуют три водородных связи более устойчивые, чем двойные это важный критерий для определения фрагмента генома). Особых различий не наблюдается. |
 |
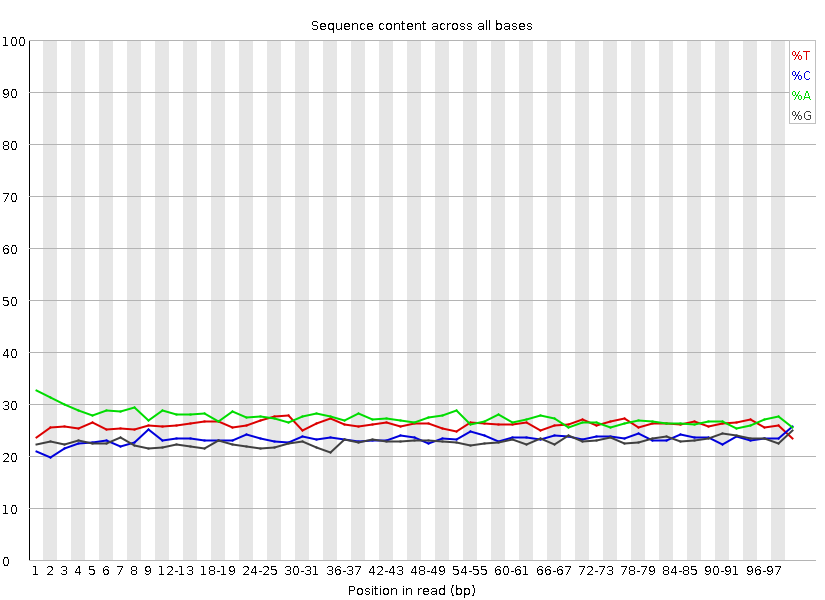 |
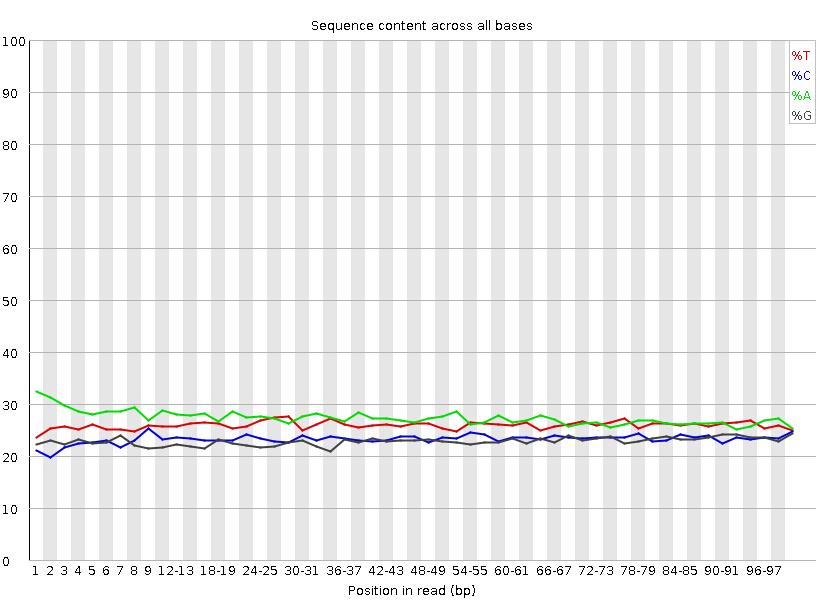
Отражает процентное содержание каждого нуклеотида в каждой позиции всех ридов. Особых различий не наблюдается. |
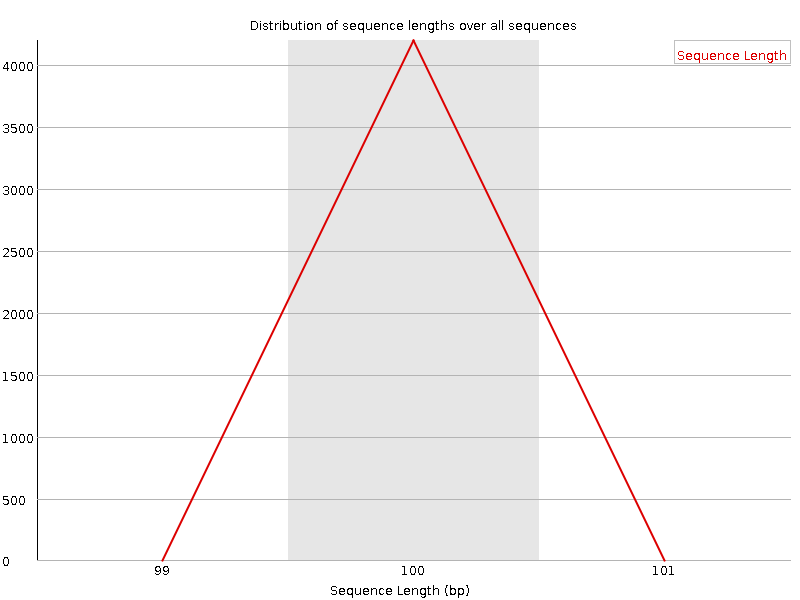 |
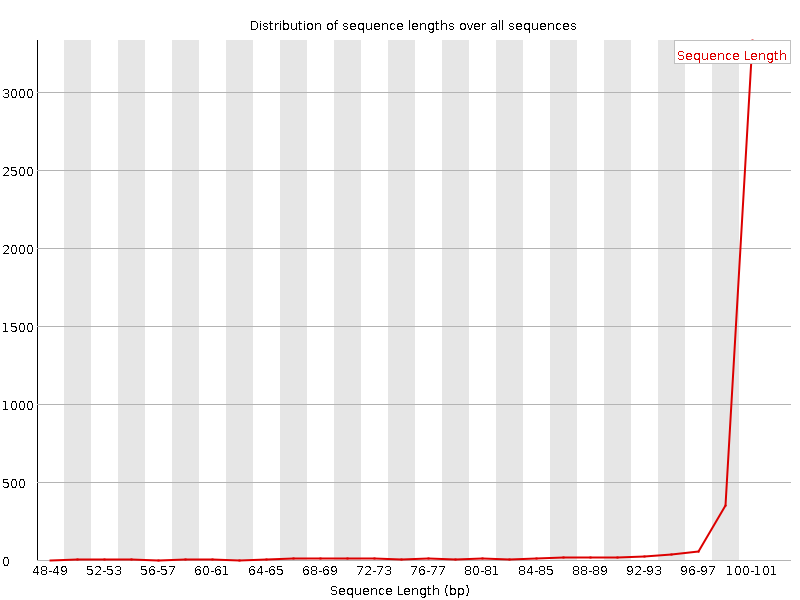 |
Распределение длин последовательностей. Очень трудно сказать, что изменилось, так как графики распределения выглядят по-разному. Судя по первой картинки до очистки не было последовательностей меньше 99,.. и больше 101,... После обработки распределение изменилось стало больше последовательностей длиной от 90 до 100. |
Картирование чтений
Сначала нам нужно создать навигацию - т.е. проиндексировать рефересную последовательность
bwa index chr11.fasta
Затем выравнять наши риды - т.е. определить положение в системе координат.
bwa mem chr11.fasta chr11_after_screen.fastq > aln.sam
Анализ выравнивания
Выравнивания в sam формате весьма громоздки. Обрабатывать их довольно долго. Поэтому переведем его в бинарный формат.
samtools view -b -o aln.bam aln.sam
Отсортируем их (выравнивания) по начальным координатам относительно рефересного генома.
samtools sort -T /ngs/anandia/aln.sorted -o aln.sorted.bam aln.bamТеперь заново проиндексируем.
samtools index aln.sorted.bam
Узнаем, сколько чтений откартировалось на геном
samtools idxstats aln.sorted.bam
В итоге 4062 последовательности откартировались, а 2 нет.
Немножко о покрытиии
Как узнать хорошее ли покрытие дпнного полиморфизма и вообще что это такое? Покрытие - это количество ридов наложившихся друг на друга при картировании.
samtools depth aln.sorted.bamЭто команда возвратила таблицу с информацией о покрытии каждого нуклеотида (диапозон значений: от 1 до 185). Выбрав нуклеотид с покрытием 181 (координата 116628504), я установила что он находится в экзоне гена BUD13, который имеет координаты: 116628482-116628666.
samtools depth aln.sorted.bam -r chr11:116628482-116628666Программа выдала понуклеотидное покрытие экзона. Среднее покрытие оказалось 143 - довольно высокий результат. Но чаще всего покрытие нуклеотида составляет 60-80.
Анализ SNP
Поиск SNP и инделей
Создадим файл с полиморфизмами.
samtools mpileup -uf chr11.fasta aln.sorted.bam > snp.bcf
Создадим файл со списком отличий между референсом и чтениями в формате .vcf.
bcftools call -cv snp.bcf > snp.vcf
Файл snp.vcf содержит 23 полиморфизма: 1 вставка, 1 делеция, 21 замена. Качество 6-ти (26%) не достигает и 30. По-моему есть некая зависимость между покрытием и качеством, так как полиморфизмф с низким покрытием имеют низкий показатель качества. Остальные полиморфизмы имеют очень высокие показатели качества. Информация о некоторых из них представлена в Таблице 1.
Таблица с информацией о трех разных полиморфизмах из 11 хромосомы
| Координата | Тип полиморфизма | Референсная последовательность | Чтение | Качество чтений | Глубина покрытия |
| 116657590 | Индель | СAAA | CAAAA | 144.467 | 63 |
| 116650454 | Индель | ATCTCT | ATCT | 196.468 | 22 |
| 116655600 | Замена | G | A | 225.009 | 37 |
Аннотация SNP
Выполняется с помощью с annovar.
Для начала удалим из vcf-файла индели, аннотация которых нас не интересует.
Затем переведем vcf файл в тот, с которым работает annovar.
perl convert2annovar.pl -format /nfs/srv/databases/ngs/anandia/snp.vcf > /nfs/srv/databases/ngs/anandia/snp.avinput
Затем последовательно были аннотированы SNP по различным базам данных.
- Refgene - база данных для метода аннотации полиморфизмов, основанного на изменении строении генов и
межгенных участков при инделях или заменах. Программа выдает 2 файла: в первом суммарная информация о всех найденных (т.е. генноразмеченных)
полиморфизмах. Там есть свои группы snp (см.рис.3). Во втором файле содержится информация о полиморфизма в экзонах. В нем тоже есть своя градация по
последствиям этих snp (см. рис.4)
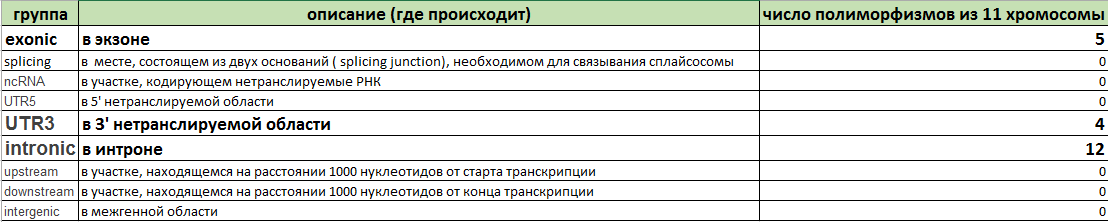
Рисунок 3 Полный список типов snp в зависимости от места их расположения.
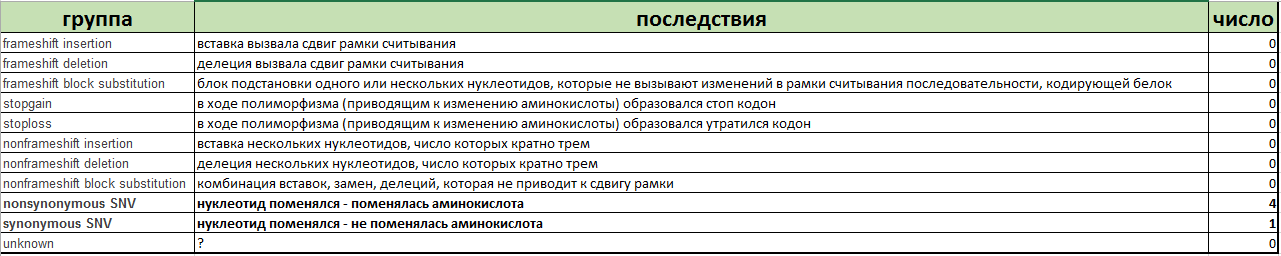
Рисунок 4 Список разных экзонных snp.
perl /nfs/srv/databases/anandia/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_refgene /nfs/srv/databases/annovar/humandb/
5 полиморфизмов были найдены внутри экзонов 3 генов.В гене KCNG11 присутствуют 2 несинонимичные замены (G748A:p.V250I; A67G:p.K23E; (т.е. замена одно нуклеотида меняет нам аминокислоту). В этом гене закодирован интегральный мембранный белок входящего АТФ-зависимого калиевого канала (закачивает ионы калия в клетку), который закрывается, когда высокая концентрация АТФ вызывает секрецию инсулина. Мутации в этом гене вызывают неконтролируемый синтез инсулина (гиперинсулинная гипогликемия), а также диабет (инсулин независимый).
В гене BUD13 есть одна синонимичная замена (G480A:p.P160P) и одна несинонимичная (C443T:p.P148L). В этом гене закодирован белок, участвующий в выведении мРНК из ядра и сплайсировании pre РНК
В гене ZPR1 1 несинонимичная замена (C791T:p.A264V). В гене закодирован белок, который содержит цинковый палец и участвует в дифференциации нейронов. Мутации могут вызывать апоптоз моторных нейронов и спинальную мышечную атрофию.
Вполне возможно, что данные полиморфизмы имеют клиническое значение. - dbsnp - база данных уже аннотированных полиморфизмов. Используется в методе фильтрации
(сравнивают полиморфизмы с раннее аннотированными: если начало, конец и собственно аллели (рефересная и чтение) точно
(важное отличие от метода разметки окружения)
совпадают - берут.
perl /nfs/srv/databases/anandia/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_snp138 /nfs/srv/databases/annovar/humandb/
Не нашли соответсвия только 2 полиморфизма. Остальные были аннотированы раньше. Т.е. имеют индентификатор в этой базе (rs). - 1000genomes - база данных полиморфизмов из 1000 геномов людей. Используется для метода фильтрации.
Можем определить, с какой частотой встречается данный полиморфизм.
perl /nfs/srv/databases/anandia/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_1000g2014oct_all /nfs/srv/databases/annovar/humandb/
Как и в прошлый раз отсеились 2 полиморфизма (те же самые) - отбор по порогу встречаемости (>= 0.05) Можно заметить, что замена пуринового основания на пиримидиновое встречается реже, чем обратная замена. - GWAS - база данных для метода разметки регионов генома. Для него (метода)
не обязательно точное понуклеотидное совпадение полиморфизмов. Программа смотрит соответсвие ранее аннотированных полиморфизмов с прочитанными,
имеющими некоторое количество нуклеотидов в окружении. GWAS - база данных аннотированных полиморфизмов, которые ассоциированы с
заболеваниями или особыми последствиями, определенными в ходе множественных геномных исследований.
perl /nfs/srv/databases/anandia/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_gwasCatalog /nfs/srv/databases/annovar/humandb/
Здесь определилсь 6 полиморфизмов. 2 ассоциированы с диабетом 2 ого типа (были предположительно заявлены в refGen аннотации), 2 были ассоциированы с увеличением количества трилицеридов, которые путешествуют по кровотоку в составе комплекса с липопротеинами. Скапливаясь в большом количестве могут образовывать препятствия кровотоку, приводя к атеросклерозу. Кроме того, триглицериды значительно повышают риск развития острого воспаления поджелудочной железы – острого панкреатита. 1 был связан с возникновением метаболитического синдрома (увеличение массы висцерального жира, снижение чувствительности периферических тканей к инсулину и гиперинсулинемия, которые нарушают углеводный, липидный, пуриновый обмен, а также артериальная гипертензия). Еще один связан с ожирением.
Все заболевания связаны с углеводным и липидным обменом. Возможно, что все эти полиморфизмы находятся либо в одном гене, либо в генах, кодирующих схожие белки. В любом случае у человекас таким набором полиморфизмов есть риск получить заболевание связанное с липидным и углеводным обменом. Интересно, сколько нужно полиморфизмов, чтобы заболевание обязательно проявилось? Делятся ли заболевание на тем, что могут с большой вероятностью проявится в связи с единичным полиморфизмов и те, которым нужна целая серия?. - Clinvar - база для метода фильтрации. Содержит информацию о связи между полиморфизмом и его влиянием на организм человека
(патогенный, неизвестный, отвечающий на лечение лекарствами и др.)
perl /nfs/srv/databases/anandia/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_Clinvar /nfs/srv/databases/annovar/humandb/
Только 2 полиморфизма были аннотированы по такой базе: один непатогенный и неспецифичный, другой - отвечающий на лечение диабет 2-ого типа.
Таблицу с информацией о всех вышеупомянутых аннотациях snp можно найти.
здесь
Таблица использованных команд
| Команда | Что делает |
| fastQC | Проверяет качество ридов. Отчет в zip архиве. |
| java -jar /usr/share/java/trimmomatic.jar SE -phred33 infile.fastq outfile.fastq TRAILING:20 MINLEN:50 | Отрезает с конца каждого чтения нуклеотиды с качеством ниже 20, оставляет только чтения длиной не меньше 50 нуклеотидов. |
| bwa index chr11.fasta | Индексирует рефересную последовательность chr11.fasta |
| bwa mem chr11.fasta chr11_after_screen.fastq > aln.sam | Строит выравнивание индексированной рефересной последовательности и ридов |
| samtools view -b -o aln.bam aln.sam | Переводит выравнивание в sam формате в бинарный. |
| samtools sort -T /ngs/anandia/aln.sorted -o aln.sorted.bam aln.bam | Сортирует bam - файл по началу в рефересном геноме |
| samtools index aln.sorted.bam | Индексирует бинарный отсортированный файл |
| samtools idxstats aln.sorted.bam | Выдает табличку с информацией о количестве откартированных на геном чтений |
| samtools mpileup -uf chr11.fasta aln.sorted.bam > snp.bcf | Создает файл с полиморфизмами |
| bcftools call -cv snp.bcf > snp.vcf | Переводит полученный .bcf файл в список отличий между референсом и чтениями в формате .vcf. |
| perl convert2annovar.pl -format /nfs/srv/databases/ngs/anandia/snp.vcf > /nfs/srv/databases/ngs/anandia/snp.avinput | Создает на основе файла .vcf файл, с которым может работать annovar |
| perl /nfs/srv/databases/anandia/annotate_variation.pl -geneanno -dbtype refGene -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_refgene /nfs/srv/databases/annovar/humandb/ | Аннотирует файл со списком SNP по базе данных refgene с использованием сборки генома человека h19 |
| perl /nfs/srv/databases/anandia/annotate_variation.pl -filter -dbtype snp138 -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_snp138 /nfs/srv/databases/annovar/humandb/ | Аннотирует файл со списком SNP по базе данных dbsnp |
| perl /nfs/srv/databases/anandia/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_1000g2014oct_all /nfs/srv/databases/annovar/humandb/ | Аннотирует файл со списком SNP по базе данных 1000genomes |
| perl /nfs/srv/databases/anandia/annotate_variation.pl -regionanno -dbtype gwasCatalog -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_gwasCatalog /nfs/srv/databases/annovar/humandb | Аннотирует файл со списком SNP по базе данных GWAS |
| perl /nfs/srv/databases/anandia/annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 /nfs/srv/databases/anandia/snp.avinput -outfile snp_annot_Clinvar /nfs/srv/databases/annovar/humandb/ | Аннотирует файл со списком SNP по базе данных Clinvar |
© 2014 Макарова Надежда