Сборка de novo
Сборка и выравнивнаие контигов с хромосомой
В предыдущем практикуме мы собирали геном, опираясь на рефересный. Но в этот раз попытаемся собрать его без подсказок. Возьмем очищенные программой Trimmomatic риды и подадим их программе velveth. Она выделит все возможные k-меры (нуклеотидные последовательности длиной k - в нашем случае k = 31).
velveth chr11_de_novo 31 -fastq -short chr11_after_screen.fastqДалее другая программа должна будет собрать k-меры в контиги, построив ориентированный граф де Брёйна.
velvetg chr11_de_novo > velvetg_dataРезультаты таковы: N50 - 266, максимальная длина контига - 1633. файл Log с информацией о N50 ; файл с последовательностями контигов
Теперь проверим результат - выравним контиги с рефересной последовательностью.
Для этого составим базу данных из последовательности хромосомы 11
makeblastdb -in chr11.fasta -dbtype nuclИ произведем поиск по ней.
blastn -db chr11.fasta -query chr11_de_novo/contig.fa -outfmt 6 -out align_contigВ результате была получена таблица , содержащая информацию о 17056 выравниваниях. При этом последовательностей query всего 113. Все последовательности, которые неоднократно наложились на 11 хромосому представлены на Рисунке 1.
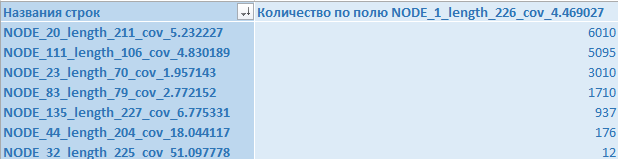
Рисунок 1. Список повторяющихся контигов с указанием количества повторов.
Таблицу с контигами можно найти здесь. На первом листе представлены все контиги, на листе Uniq_contig представлены однократно встречающиеся. Всего контигов, которые единственным образом картируются на хромосому 107. Они расположены по возрастанию начальной по прямой цепи координате. Отрицательным значениям в последней колонке соответсвуют перекрытия, положительным - разрывы.
Итак, вычислив величину разрывов и перекрытий у контигов было установлено, что друг за дружкой легли контиги только в двух местах, встретилось 82 перекрытия и 22 разрыва.
Анализ полученных результатов
Контиги, которые многократно легли на последовательность хромосомы
Предположительно, повторы могут являться мобильными элементами.
Я взяла самый повторяющийся контиг - NODE_20_length_211_cov_5.232227. Был осуществлен поиск blastn по базе human genomic plus transcript.
Результаты представлены на Рисунке 2. Последовательность контига найдена и сборке hg38 (самая новая) и в CHM1_1.1.
Самое интересное, что последовательность контига также найдена в траскрипте гена кальциевого канала
(который обсуждался в прошлом практикуме относительно полиморфизмов).
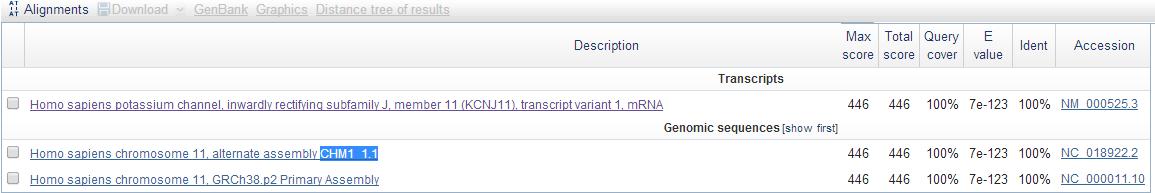
Рисунок 2. Находки megablast относительно контига NODE_20_length_211_cov_5.232227.
Далее я посмотрела встречаемость данной последовательности во всем геноме человека. Как минимум 20000 находок (максимальная выдача blast) было определено.
Все были с хорошим E-value (min 2e-58) и процентом идентечности (min 78 %). Важно заметить, что среди выдачи были последовательности траскриптов.
Правда было их не очень много (576). Особое внимание следует уделить находкам с необычным названием ALU (76 находки).
Теперь посмотрим на участок 11 хромосомы, где находится данный повтор, с помощью UCSC Genome Browser. Структура представлена на Рисунке 3.
Мы можем увидеть две изоформы белка кальциевого канала. Только в одном варианте транскрипта экзона (более длинном) есть наш повтор.
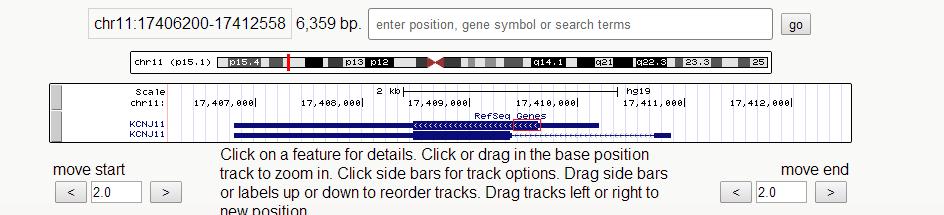
Рисунок 3. Изображение структуры гена KCNJ11 в UCSC Genome Browser. Участок повтора (координаты: 17409608 - 17409848) выделен красным прямоугольником.
Итак, что у нас в итоге: длина повтора 250, встречается в транскриптоме, влияют на сплайсинг, некоторые ассоциированы с раком (находки с Alu), не содержит кодирующих последовательностей (своих собственных белков). Вывод: данный повтор является распространенным ретротранспозоном, встречающимся в геноме человека. Их называют ALU и они образуют целое семейство. Alu, встраиваясь в ген, может вносить в него дополнительные сайты сплайсинга, увеличивая разнообразие считываемых с гена изоформ мРНК. Про него можно узнать здесь
Перекрытия
Почему при существовании перекрытий программа не объединила несколько контигов в один?
-
Первая причина состоит в том, что внутри перекрытия есть различия в один или несколько нуклеотидов (см. Рисунок 4). Это происходит из-за ошибок во время секвенирования. Это и не дает программе соединить два контига в один.
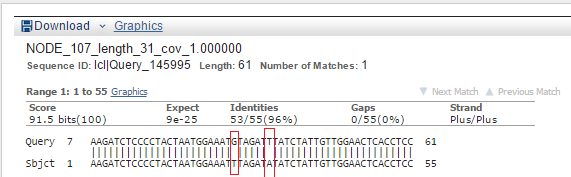
Рисунок 4. Выравнивание двух контигов (NODE_109_length_31_cov_1.000000 и NODE_107_length_31_cov_1.000000 ) с перекрытием 54 нуклеотида. Несоответсвия нуклеотидов выделены красными прямоугольниками. Очень много встречается перекрытий длиной 29. Это меньше длины k-мера (31). И этого недостаточно, чтобы продолжить граф де Брайна (ребра, которого составляют k-меры).
Разрывы
Таблица с выравниванием одиночных контигов отсортирована по началу в геноме по прямой цепи. В этой таблице выделяются две области (по координатам): 17406855-17409834, и 116618794-116658814. Т.е. получается, что мы секвенировали не весь геном, и даже не всю хромосому, а конкретный участок, содержащий определенный ген (KCNJ11 - тот самый, мутации в котором вызывают нарушение обмена углеводов и липидов, приводя к диабету и атеросклерозу) и (BUD13 и ZPR1). Вот плчему полиморфизмы были найдены только в этих генах. Т.к. секвенирование генома человека имеет медицинские цели, то и секвенируют только то, что необходимо для диагностики (необходимые экзоны). Почти все разрывы, которые попадались, были либо в интронах, либо в межгенной области. Намеренный поиск выявил разрыв (53 нуклеотида) внутри гена KCNJ11 - см. Рисунок 5. Объяснение: вероятно ошибка программы.
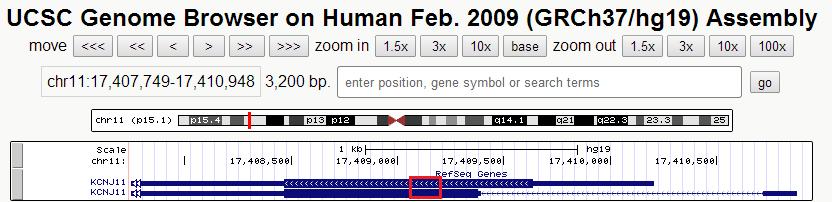
Рисунок 5. Участок гена KCNJ11, в котором обнаружен разрыв между контигами NODE_10_length_331_cov_7.039275 и NODE_12_length_72_cov_3.305556.
Контиги на той же цепи, что и ген. (были случаи, когда на противоположной гену цепи есть разрывы между контигами). Координаты разрыва: 17409075 - 17409128.
Зависимость качества сборки от длины k-меров
Последовательно были запущены velveth и velvetg c указанием k-мера меньшей длины. Результаты представлены в таблице. Самая лучшая сборка из всех представленных получится при k=29 (возможно, это связано с тем, что много перекрытий было длиной 29), а худшая при k=27. Так как чем лучше N50, тем лучше сборка.| Длина k-мера | N50 | Максимальная длина контига | Качество по сравнению с k-31 |
| 31 | 266 | 1633 | |
| 29 | 306 | 1635 | улучшилось |
| 27 | 263 | 913 | ухудшилось |
| 25 | 279 | 928 | ухудшилось |
| 19 | 276 | 928 | ухудшилось |
© 2014 Макарова Надежда