Была рассмотрена последовательность ДНК прочтенная ранее (в 6 практикуме) на основании данных, полученных из капиллярного секвенатора. Последовательность в формате fasta:
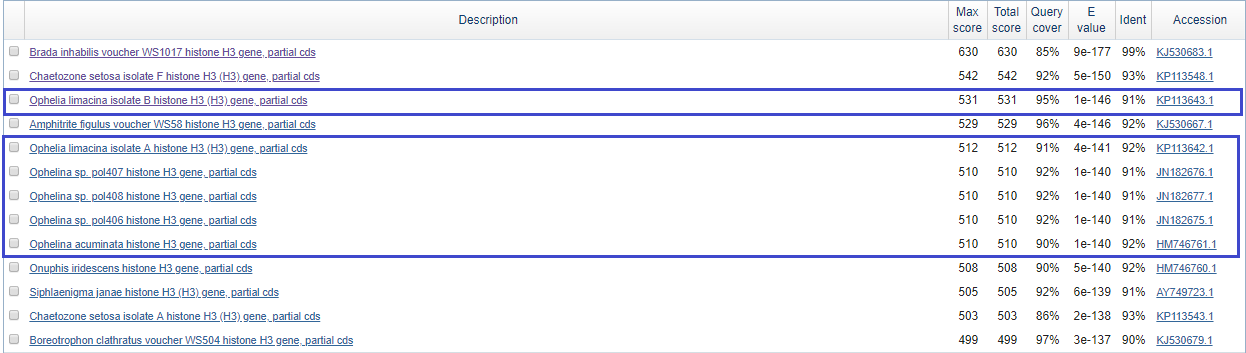
прямая цепь, отображенная обратная цепь. Из двух прочтений с помощью программы consambig пакета EMBOSS была сгенерирована консенсусная последовательность.Изображение 1: основные гомологи, найденные BLASTN (упорядочены по убыванию счета выравниявания).
Сначала я рассмотрела три находки с наибольшим счетом и три организма частью генома которых эти находки являются, последовательно: Brada inhabilis, Chaetozone setosa и Ophelia limacina.

Изображение 2:слева-напрво Brada inhabilis, Chaetozone setosa и Ophelia limacina.
Эти три организма принадлежат к кольчатым червям (Annelida), к классу многощетинковые черви (Polychaeta), поэтому с большой
долей вероятности можно сказать, что исследуемый фрагмент ДНК является участком генома полихеты.
Среди находок с наибольшим
счетом чаще всего встречаются представители рода Ophelia (Изображение 1, обведены синим), но среди трех гомологов с наибольшим счетом выравнивания фрагмент, принадлежащий Ophelia limacina,
имеет наименьший счет и процент идентичности, поэтому однозначно отнести изучаемый фрагмент к этому роду нельзя. Далее я осортировала результаты
поиска по убыванию процента идентичности.
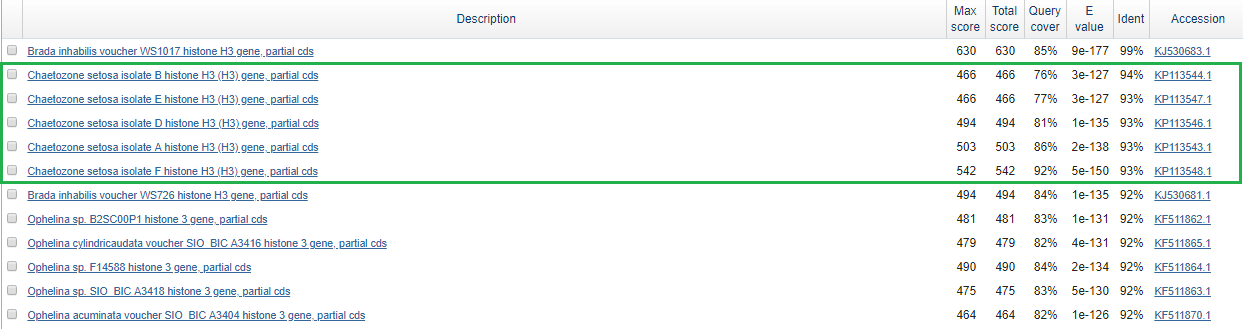
Изображение 3: основные гомологи, найденные BLASTN (упорядочены по убыванию процента идентичности).
Такой порядок мне кажется более показательным, потому что, как видно из изображения 4, данные находки имеют меньший счет при большом проценте
идентичности из-за несовпадений выравниваний в конце и/или в начале цепи. Но в связи с тем, что секвенирование не дает точных результатов для начала и
конца исследуемого участка, а также с тем, что секвенируемый участок мог быть поврежден (оборван в начале или в конце), идентичность в середине фрагмета имеет большую
значимость, чем идентичность в концевых участках. Выравнивания с фрагментами взятыми у представителей рода Ophelina имеют
сравнительноменьший процент идентичности, поэтомуданный родяизрассмотрения исключила.

Изображение 4: основные гомологи, найденные BLASTN (упорядочены по убыванию процента идентичности).
Большое количество участков генома, имеющих высокий процент идентичности, принадлежит Chaetozone setosa (Изображение2: выделены желтым). Однако процент идентичности
Brada inhabilis все же ощутимо выше. Выравнивание исследуемого фрагмета с гомологом, принадлежащим Brada inhabilis все же ощутимо выше. Было рассмотрено выравнивание с фрагментом генома Brada inhabilis (изображение 5). Из данноговыравнивания
видно, что фрагменты имеют всего один несовпадающий нуклеотид, напротив цитозина Brada inhabilis в той позиции, где у исследуемого фрагмента наблюдается полиморфизм аденина и гуанина, то есть наблюдается частичное совпадение. В результате можно
с большой долей вероятности сказать,что исследуемый фрагмент принадлежит геному Brada inhabilis.
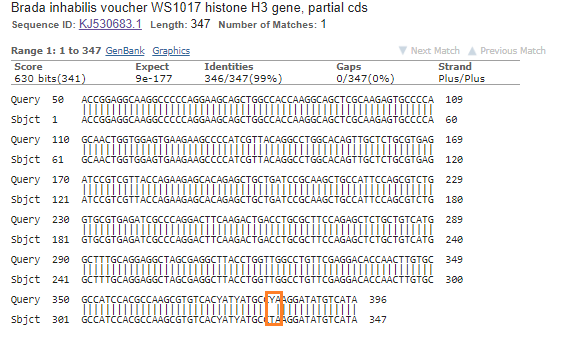
Изображение 5: выравнивание исследуемого фрагмета с гомологом, принадлежащим Brada inhabilis.
Так как совпадение с геном Brada inhabilis почти полное я предпологаю, что данные для соответствующего учатка генома Brada inhabilis
верны и для исследуемого фрагмента ДНК, в томчисле исследуемый фрагмент кодирует белок "histone H3"(как и соответствующий
участок генома Brada inhabilis).
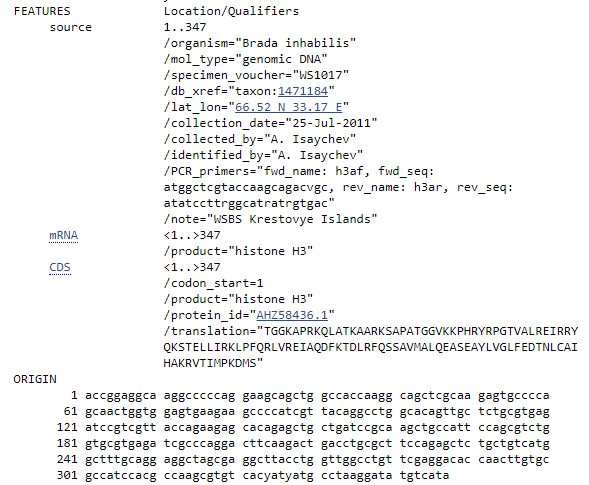
Изображение 6: данные об участке генома Brada inhabilis предположительно верные и для исследуемого фрагмента.
Для сравнения трех вариантов алгоритмов blast (megablast, blastn с параметрами по умолчанию и blastn с максимально чувствительными параметрами)
был произведен поиск гомологов сначало для консенсусной последовательности из предыдущего задания, а затем для
последовательность некодирующей РНК митохондриального генома Beroe forskalii (AC митохондриального генома:MG655622.1), а именно участок MG655622.1:1880..2255:rRNA(Beroe forskalii 12S ribosomal RNA).
Замечания к поиску гомологовдля ДНК из 1 задания:При поиске без ограничения таксона 3 алгоритма показывали почти
одинаковые результаты(совпадающийе соответственно с описанными в задании один,
поэтому мне стало инстересно, как справится каждый алгоритм, если убрать "основного претендента" и поискать гомологов в других таксонах, поэтому
я исключила из поиска таксон Annelida (для всех 3 алгоритмов). Порог e-valeu для всех 3 алгоритмов был установлен 5e-130.
Я дополнительно увеличила штраф за несовпадение в blastn с максимально чувствительными параметрами, чтобы сделать
его не только чувствительным, но и максимально точным.
Замечания к поиску гомологовдля RNK из 2 задания:порог e-valeu для всех 3 алгоритмов был установлен 0.1.
Таблица 1:Сравните списков нуклеотидных последовательностей найденных тремя разными вариантами blast.
Название алгоритма |
megablast |
blastn с параметрами по умолчанию |
blastn с максимально чувствительными параметрами |
|---|---|---|---|
Основные параметры запуска (для ДНК из задания 1) |
Длина слова:24 Match/Mismatch Scores:1,-2 Gap Costs:linear |
Длина слова:11 Match/Mismatch Scores:2,-3 Gap Costs:5,2 |
Длина слова:7 Match/Mismatch Scores:1,-3 Gap Costs:5,2 |
Суммарное количество находок (для ДНК из задания 1) |
11 |
98 |
1 |
Основные параметры запуска (для рРНК) |
Длина слова:28 Match/Mismatch Scores:1,-2 Gap Costs:linear |
Длина слова:11 Match/Mismatch Scores:2,-3 Gap Costs:5,2 |
Длина слова:7 Match/Mismatch Scores:1,-3 Gap Costs:5,2 |
Суммарное количество находок (для рРНК) |
3 |
99 |
5 |
Сравнение алгоритмов при первом поиске: то, что результаты работы каждого алгоритма сильно отличаются видно уже из количества находок. blastn с максимально чувствительными параметрами и, установленными мною, большими штрафами
за несовпадение нашел всего 1 гомолог, но очень "качественный":Siphlaenigma janae histone H3- ген, кодирующий белок гистона Siphlaenigma janae (поденка). Как я выяснила в задании 1
исследуемый ген у червей также histone H3. Гены двух организмов ,имеющие одинаковые функции, имеют очень схожее строение, это дает нам возможность предположить, что эти два организма эволюционно родственны.
Больше всего гомологов было найдено blastn с параметрами по умолчанию, при чем лучшей находкой является histone H3 ген Boreotrophon clathratus(морская улитка), когда ген Siphlaenigma janae занимает только второе
место(это связано скорее не с длиной слова, а с меньшим штравом за несовпадение).
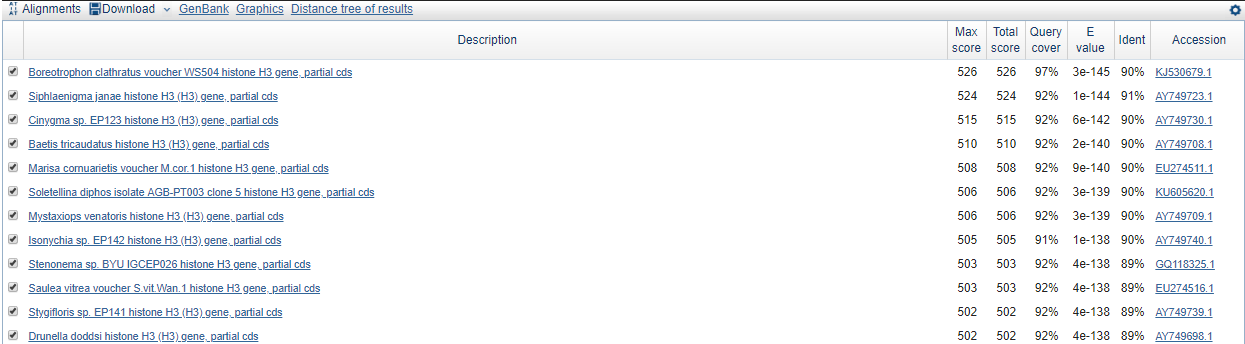
Изображение 7: основные гомологи ДНК, найденные BLASTN c параметрами по умолчанию.
Сравнение алгоритмов при втором поиске:так как я искала по самой последовательности рРНК (а не по AC) на первом месте среди гомологов во всех 3 поисках стоит сама эта послндовательность и процентом
идентичности 100%. А вот дальшее результаты работы 3 алгоритмов значительно различаются: blstn с параметрами по умолчанию нашел очень много бактериальных геноd, megablast выдал только митохондриальные гены того же вида гребневиков, а blastn с более чувствительными параметрами дал на выходе
митохондриальные гены гребневика и еще 2 гена бактерий (по всей видимости наиболее близких к гену гребневика).
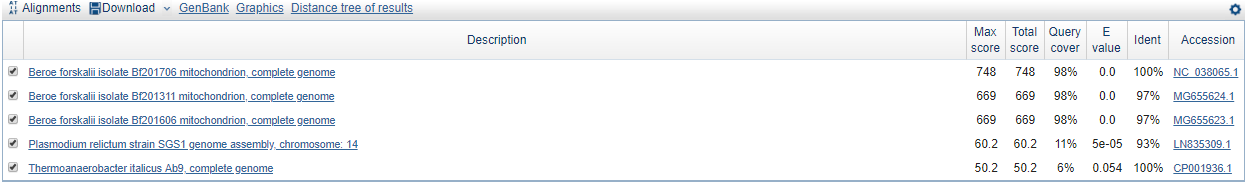
Изображение 8: основные гомологи рРНК, найденные blastn с максимально чувствительными параметрами.
Было проверено наличие гомологов трех белков в неаннотированном геноме Amoeboaphelidium protococcarum (примитивный родственник грибов).
Поиск производился с помошью BLAST+ на kodomo.
HSP71_YEAST, шаперон HSP70, белок теплового шока: есть хорошее совпадение эталонного гена с участком scaffold-199, счет выравнивания 948, e-valeu стремится к 0. Однозначно можно сказать, что такой ген в геноме есть.
HIS31_HUMAN, гистон H3, белок, участвующий в образовании нуклеосомы:есть два гомолога, выравнивания с каждым из которых имеют одинаковый счет(250), а именно scaffold-104 и unplaced-368. Качество выравнивания
и двойственность совпадения не дает возможности однозначно судить о наличии гена, кодирующего данный белок в геноме.
PRPC_EMENI, митохондриальная цитратсинтаза: нет достоверных совпадений, лучшее выравнивание захватывает только малую часть эталонного гена, поэтому такого гена в геноме, скорее всего нет, или же он сильно видоизменен.
Для рассмотрения был выбран scaffold-420, участок генома Amoeboaphelidium protococcarum длиной 73285 нуклеотидов. Поиск генов
производился с помощью blastn на сайте NCBI, поиск только внутри таксона Fungi, по алгоритму megablast с параметрами по умолчанию.
Megablast нашел несколько сходных генов разных видов грибов в одном и том же месте исследуемого участка, среди них много фрагментов с неаннотированными функциями, но есть и ген
с достаточно высоким процентом идентичности (80) и счетои (228 бит), кодирующий 60S рибосомальный белок L4 (TDEL0D03920) у Torulaspora delbrueckii.
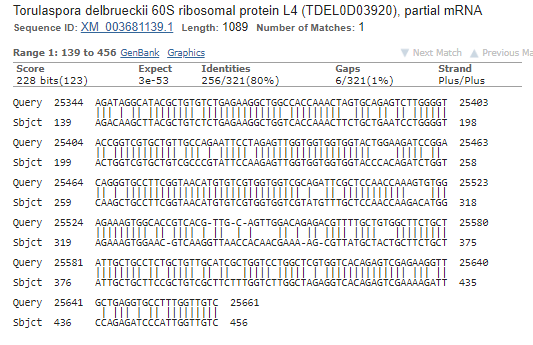
Изображение 9:выравнивание участка scaffold-51 с геном Torulaspora delbrueckii.
Соответствие не полное, но все же значительно выше случайного, а значит вполне
можно предположить, что scaffold-420 содержит в себе ген, кодирующий одну из субъединиц рибосом Amoeboaphelidium protococcarum.
Я выбрала род Dictyoglomus, в нем всего 2 вида Dictyoglomus thermophilum (типовой вид) и Dictyoglomus turgidum. Dictyoglomus thermophilum является экстремально термофильной (оптимальная температура 73 °C). Для обоих видов доступны полные геномы на NCBI, соответственно их и буду рассматривать. По алгоритму megablast была построена карта локального сходства.
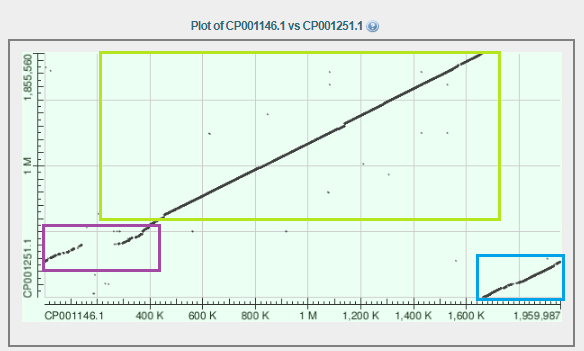
Изображение 10:карта локального сходства для Dictyoglomus thermophilum (по вертикали) и Dictyoglomus turgidum(по горизонтали).
По данной карте однозначно можно определить, что она построена для близкородственных организмов. Голубым выделен крупный гомологичный участок. Участок выделенный зеленым
также соответствует гомологичный фрагментам генома, он не встал "в общую линию", но в данном случае надо помнить, что бактерии имеют одну кольцевую молекулу ДНК. Данный участок при развертывании и прочтении просто оказался для одной бактерии в
начале генома, а для другой в конце. Фиолетовым выделен участок карты, соответствующий участкам генома, претерпевшим значительные изменения при расхождении данных видов. Именно этот участок позволяет сказать, что геномы по которым была
построена карта все же не принадлежат организмам одного вида.
Третий семестр(осенний семестр 2018)