Суть задания состоит в расчёте точечных зарядов на атомах этана. Построение файла топологии, этот файл содержит описание ковалентных и нековалентных взаимодействий. С помощью расчёта энтальпии испарения предлагается найти оптимальные параметры для описания Ван-дер-Ваальсовых взаимодействий.
Начнем с того, что определим точечные заряды. Для этого воспользуемся набором скриптов RED на perl.
С помощью babel сделаем pdb файл этана из результатов оптимизации из предыдущего практикума:
et.pdb
Добавим путь к скриптам в системный путь:
export PATH=${PATH}:/home/preps/golovin/progs/bin
Теперь с помощью скрипта Ante_RED.pl подготовим pdb файл.
Ante_RED.pl et.pdb
Переименуем p2n файл в Mol_red1.p2n (входной файл следующего скрипта). Запускаем RED:
RED-vIII.4.pl
В директории Data-RED находим файл Mol_m1-o1.mol2 с координатами атомов и зарядами.
Начнем создание файла описания молекулы в формате пакета программ GROMACS. Единица измерения расстояния в GROMACS
нанометр.
Итак первые две строчки, здесь мы задаём некоторые правила:
[ defaults ] ; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ 1 2 yes 0.5 0.8333
Дальше мы задаём типы атомов и собственно параметры для функции Ленорда-Джонса. Будем считать, что в случае этана Ван-дер-Ваальсовое взаимодействие между атомами углерода разных молекул минимально, так как углероды почти полностью экранированы атомами водорода. Поэтому поставим для углерода некоторые параметры. Ван-дер-Ваальсовый радиус водорода, т.е. сигма нам известен из многих источников, см. webelements.com. Итак у нас получилось, что в этом разделе всего одна переменная это epsilon для водорода.
[ atomtypes ] ; name at.num mass charge ptype sigma epsilon H 1 1.008 0.0000 A 1.06908e-01 1.00000e-00 C 6 12.01 0.0000 A 3.39967e-01 3.59824e-01
Дальше переходим непосредственно к описанию молекулы. Здесь мы описываем имя и указываем, что соседи через три связи не учитываются при расчете Ван-дер-Ваальсовых взаимодействий. Это верно так, как мы включаем это взаимодействие в торсионные углы:
[ moleculetype ] ; Name nrexcl et 3
Добавим атомы этана. В первом столбце идёт номер атома. На него мы будем ссылаться при описании связей.
[ atoms ]
; nr type resnr residue atom cgnr charge mass
1 C 1 ETH C1 1 -0.0189 12.01
2 C 1 ETH C2 2 -0.0155 12.01
3 H 1 ETH H1 3 0.0059 1.008
4 H 1 ETH H2 4 0.0059 1.008
5 H 1 ETH H3 5 0.0059 1.008
6 H 1 ETH H4 6 0.0056 1.008
7 H 1 ETH H5 7 0.0056 1.008
8 H 1 ETH H6 8 0.0056 1.008
Переходим к описанию связей. Константу жесткости и длину связи берем из занятия 4.
[ bonds ]
; ai aj funct b0 kb
1 2 1 0.1554 150000.0
1 3 1 0.1085 180000.0
1 4 1 0.1085 180000.0
1 5 1 0.1085 180000.0
2 6 1 0.1085 180000.0
2 7 1 0.1085 180000.0
2 8 1 0.1085 180000.0
Описание углов, тоже самое. Силовые константы берем из примера.
[ angles ]
; ai aj ak funct phi0 kphi
;around c1
3 1 4 1 109.500 200.400
3 1 5 1 109.500 200.400
4 1 5 1 109.500 200.400
3 1 2 1 109.500 400.400
4 1 2 1 109.500 400.400
5 1 2 1 109.500 400.400
;around c2
1 2 6 1 109.500 400.400
1 2 7 1 109.500 400.400
1 2 8 1 109.500 400.400
6 2 7 1 109.500 200.400
6 2 8 1 109.500 200.400
7 2 8 1 109.500 200.400
Торсионные углы, параметры из примера:
[ dihedrals ]
; ai aj ak al funct t0 kt mult
3 1 2 6 1 0.0 0.62760 3
3 1 2 7 1 0.0 0.62760 3
3 1 2 8 1 0.0 0.62760 3
4 1 2 6 1 0.0 0.62760 3
4 1 2 7 1 0.0 0.62760 3
4 1 2 8 1 0.0 0.62760 3
5 1 2 6 1 0.0 0.62760 3
5 1 2 7 1 0.0 0.62760 3
5 1 2 8 1 0.0 0.62760 3
Теперь создадим список пар атомов которые не должны считаться при расчете VdW.
Здесь можно возразить: а как же nrexcl=3 ? . Особенность расчета 1-4 взаимодействий подразумевает, что в
профиле торсионного угла участвует не только потенциал с cos, но и LJ отталкивание. Это удобно для точной
параметризации, но нам пока не надо. Итак добавляем список:
[ pairs ] ; ai aj funct 3 6 3 7 3 8 4 6 4 7 4 8 5 6 5 7 5 8
Итак основное описание молекулы создано. Теперь переходим к описанию системы. Используем уже готовые координаты с 38 молекулами этана. Давайте укажем это в описании:
[ System ] ; any text here first one [ molecules ] ;Name count et 38
В итоге получаем файл et.top
Итак мы создали описание молекулы с нуля. Чаще для этого используются программы с готовыми блоками.
Наша следующая задача промоделировать испарение этана. Нам любезно подготовлены два файла, описывающие два состояния системы, первое соответствует газовой фазе, где расстояния между молекулами порядка 50 ангстрем. Файл для газа. Вторая система имеет такую же плотность как и жидкий этан. Файл для жидкой фазы. Наша задача провести короткое моделирование динамики каждой из этих систем о определить разницу в энергии VdW взаимодействий между системами. И сравнить эту разнице с энтальпией испарения этана. При Т=25 это значение равно 5.4 кДж/моль. Вспомним, что epsilon для водорода нам не известна. Давайте по аналогии с занятием 4 создадим 7 топологий с разными значениями epsilon. Будем использовать скрипт:
На основе данных файлов txt получаем:
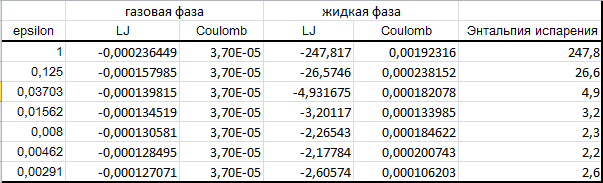
Из таблицы четко видно, что энергия VdW вносит во взаимодействие гораздо больший вклад, чем кулоновские силы. Наиболее близкое значение энергии испарения наблюдается при epsilon=0,03703. Чтобы воспроизводилась энтальпия испарения этана epsilon водорода должна лежать в диапазоне от 0.0125 до 0.03703.