Все файлы по данному практикуму находятся в папке Practice6
Изучим как реализован контроль температуры в молекулярной динамике на примере GROMACS. Объект исследования это одна молекула этана.
Начнем с того, что подготовим файл координат и файл топологии. В прошлом занятии нам был предоставлен gro файл с 38 молекулами этана. Создадим индекс файл котором будет группа из одной молекулы этана.
make_ndx -f box_38.gro -o 1.ndxПосле запуска команды появилось приглашение к вводу. Сначала ознакомимся с помощью нажав "h" + enter. Выберем остаток номер 1. Нажмите enter и увидим, что появилась новая группа.
Теперь создадим gro файл с одной молекулой и зададим ячейку . При запуске ediconf выберите номер соответствующей группе из одной молекулы.
editconf -f box_38.gro -o et1.gro -n 1.ndx #зададим ячейку и расположим молекулу по центру ячейку editconf -f et1.gro -o et.gro -d 2 -c
Получили файл et.gro
Исправим файл топологии et.top из прошлого задания. В разделе [ molecules ] изменили количество молекул этана.
Нам даны 5 файлов с разными параметрами контроля температуры:
be.mdp - метод Берендсена для контроля температуры.
vr.mdp - метод "Velocity rescale" для контроля температуры.
nh.mdp - метод Нуза-Хувера для контроля температуры.
an.mdp - метод Андерсена для контроля температуры.
sd.mdp - метод стохастической молекулярной динамики.
Начиная с этого момента можно написать скрипт по работе с 5ю системами:
Скачаем файлы при помощи скрипта.
Сначала построим входные файлы для молекулярно-динамического движка mdrun с помощью grompp:
grompp -f ${i}.mdp -c et.gro -p et.top -o et_${i}.tpr
# где i: be,vr,nh,an,sd см. выше список mdp файлов
Задать i вне скрипта можно командой export i="be".
Должно получиться 5 tpr файлов. Теперь для каждого из них запустим mdrun.
mdrun -deffnm et_${i} -v -nt 1
Для каждой из 5 систем проведем конвертацию в pdb и просмотрим в PyMol.
trjconv -f et_${i}.trr -s et_${i}.tpr -o et_${i}.pdb
- et_an.pdb - молекула колеблется относительно одного положения, незначительные колебания связей и углов, вращения нет.
- et_be.pdb - сначала наблюдаются колебания связей, потом их вращение с увеличивающейся скоростью, молекула приходит в движение и начинает вращаться.
- et_nh.pdb - наблюдается вращение метильных групп относительно С-С связи и небольшие колебания валентных углов.
- et_sd.pdb - молекула спонтанно меняет конформацию и положение в пространстве.
- et_vr.pdb - наблюдаются смены конформаций и колебания длин связей и валентных углов со значительной амплитудой, молекула большую часть времени находится в заторможенной конформации.
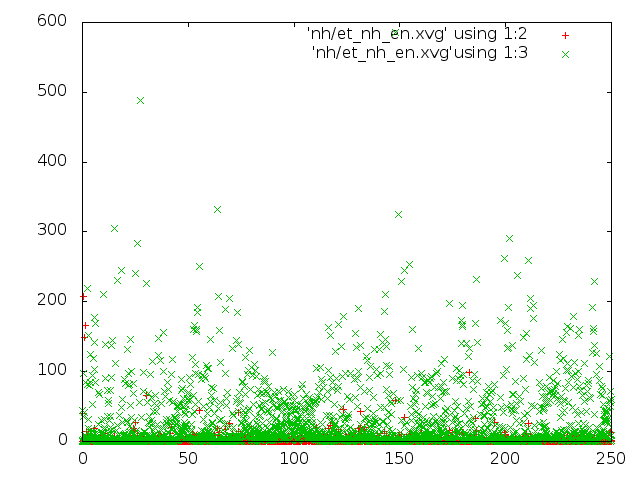
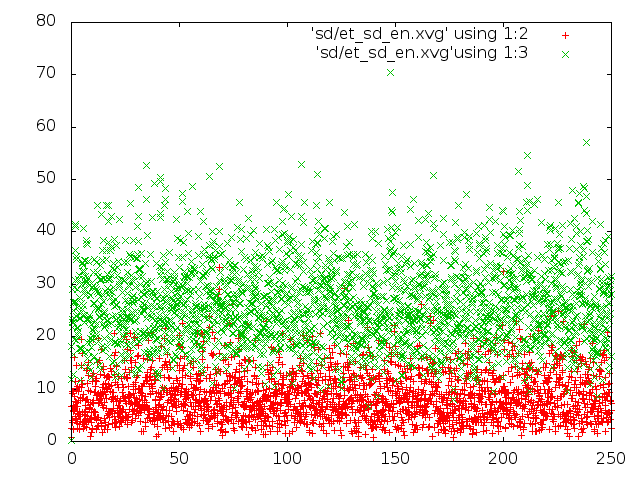
Сравним потенциальную энергию связи и кинетическую энергию для каждой из 5 систем.
g_energy -f et_${i}.edr -o et_${i}_en.xvg
Построим графики изменения энергий. При использовании Gnuplot для построения графиков,
перейдем в рабочую директорию и запустим Gnuplot:
set datafile commentschars "#@&" plot "./et_be_en.xvg" using 1:2, "./et_be_en.xvg" using 1:3 .... plot "./et_sd_en.xvg" using 1:2, "./et_sd_en.xvg" using 1:3Рассмотрим распределение длинны связи С-С за время моделирования. Сначала создадим индекс файл с одной связью. В текстовом редакторе создаем файл b.ndx со следующим содержимым:
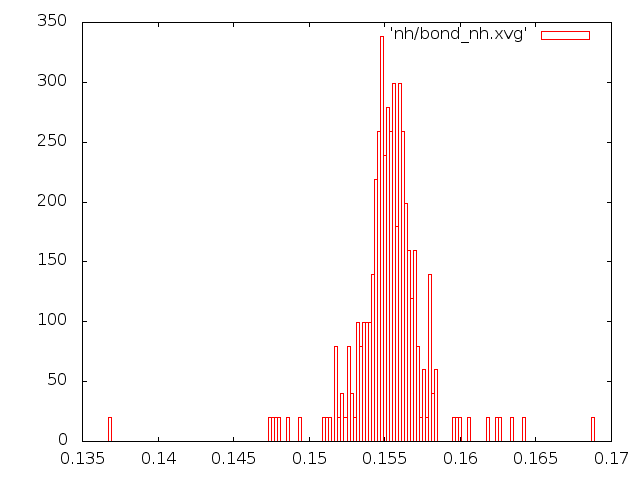
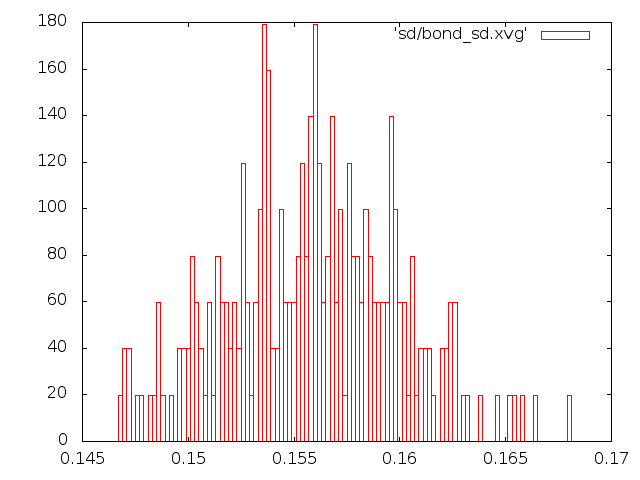
[ b ] 1 2И запустим утилиту по анализу связей g_bond:
g_bond -f et_${i}.trr -s et_${i}.tpr -o bond_${i}.xvg -n b.ndx
Построим графики распределения длин связей. Рекомендуемый вид это гистограмма
или boxes в Gnuplot.
Чтобы не делать вышеописанные операции вручную был написан скрипт.
На рисунке слева - энергия для каждого состояния, справа - распределение длин связей:
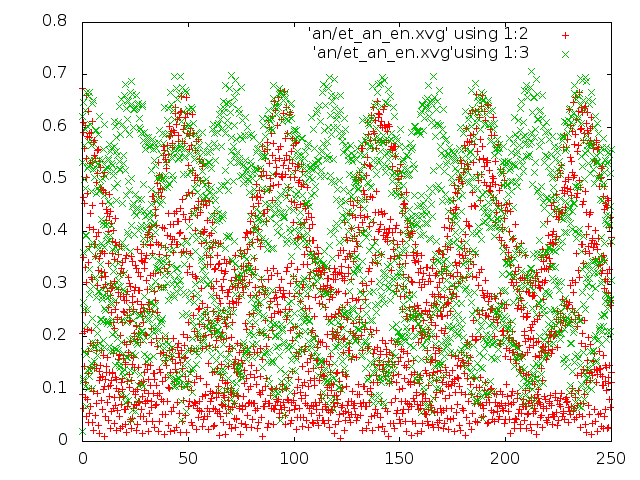
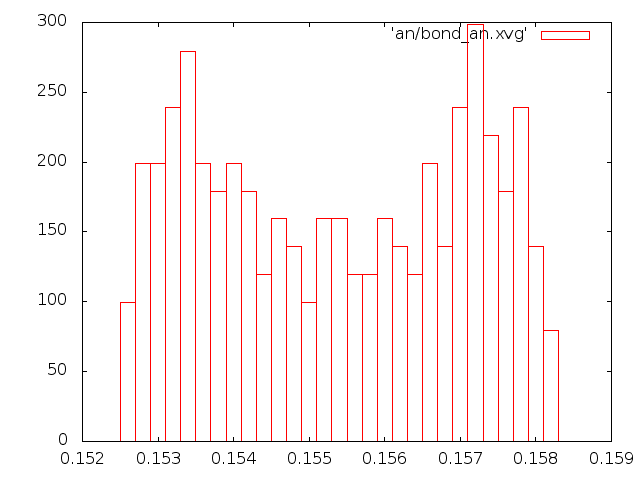
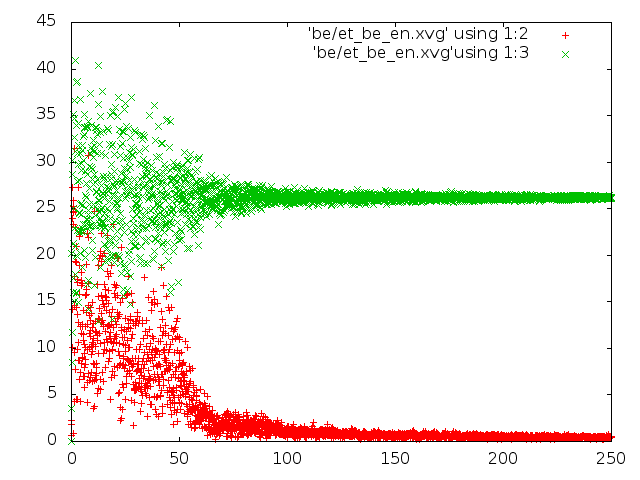
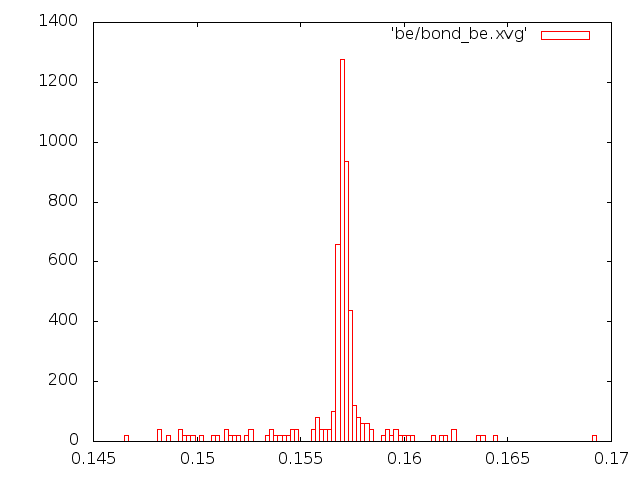
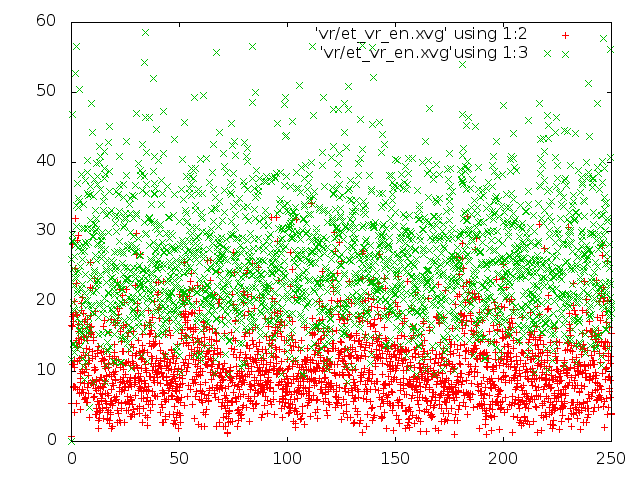
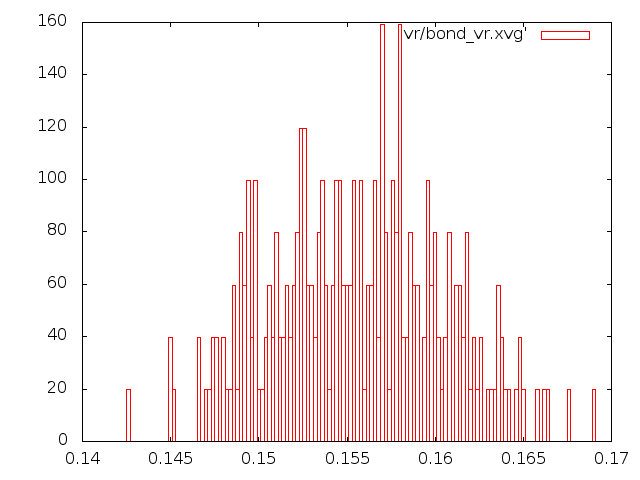
Рассмотрим распределение Максвелла-Больцмана:
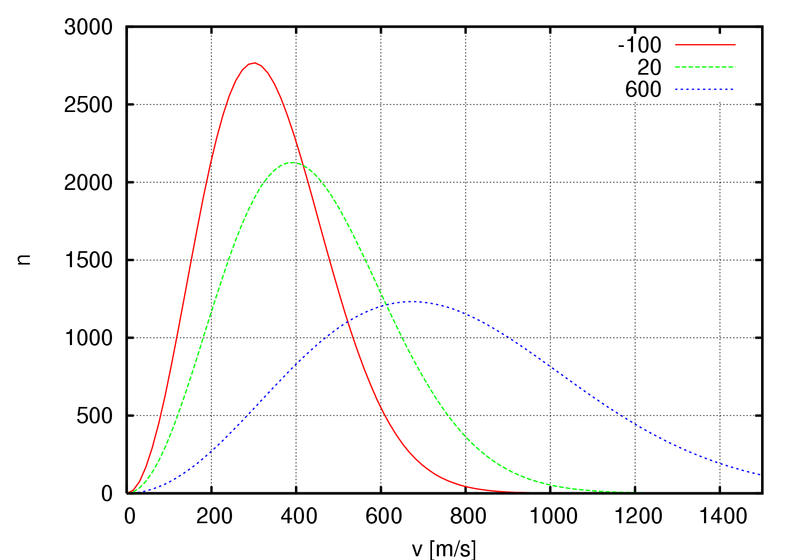
Из пяти представленных графиков на распределение Максвелла-Больцмана похожи графики, полученные стохастическим методом и методом Velocity rescale. Однако по данным PyMol из этих двух методов Velocity rescale выглядит наиболее адекватным по сравнению с хаотическим движением молекулы, полученном стохастическим методом. Таким образом, Velocity rescale кажется более подходящим для контроля температуры.