| Учебный сайт Екатерины Швецовой | |||
| Главная | Обо мне | Семестры | Ссылки |
| 1 семестр | 2 семестр | 3 семестр | 4 семестр | 7 семестр | 8 семестр |
Предсказание вторичной структуры тРНК из файла 1GTR.pdb
Работа производилась с файлом 1GTR_rna.fasta, содержащим последовательность заданной тРНК (цепь В белка 1GTR). Файл был скачан из сайта PDB.
Предсказание вторичной структуры тРНК путём поиска инвертированных повторов
Для предсказания вторичной структуры тРНК была использована программа einverted из пакета EMBOSS (ищет инвертированные повторы). Известно, что программа не работает с нестандартными нуклеотидами, но таких в моей последовательности нет, поэтому никаких замен произведено не было. В процессе варьировались значения gap penalty и minimum score threshold. В результате при gap penalty = 12 и minimum score threshold = 15 (и меньше) был найден один инвертированный повтор, соответствующий акцепторному стеблю:
SEQUENCE: Score 18: 6/6 (100%) matches, 0 gaps
1 ggggta 6
||||||
69 ccccat 64
При больших значениях minimum score threshold и том же значении gap penalty программа не находила нужных последовательностей. При других ненулевых значениях gap penalty и значении minimum score threshold меньше 15 находился только один стебель (при очень низких значениях, содержащий гэпы). При понижении gap penalty до 0 было получено следующее:
SEQUENCE: Score 72: 24/24 (100%) matches, 20 gaps
1 ggggt------a-tcgccaag---cggtaag-gcaccgg-attc 32
||||| | ||| || ||| || | |||| | ||
69 ccccatgctcctaagc--tt-ggagcc-tt-ac--ggcctt-ag 34
В найденном стебле очень много гэпов, поэтому использовать его для предсказания структуры тРНК невозможно.
Данная программа учитывает только канонические взаимодействия, поэтому поиск стеблей производит не слишком эффективно.
Предсказание вторичной структуры тРНК по алгоритму Зукера
Алгоритм Зукера реализует программа mfold. Я пользовалась online-версией. Программа запускалась несколько раз, при этом варьировался параметр Р (percent suboptimality), он указывает, на сколько процентов выдаваемое предсказание структуры может отличаться по своей вычисленной энергии от оптимального. Чем больше значение этого параметра, тем больше вариантов предсказания было на выходе. При Р=5 была найдена 1 структура с 3-мя стеблями (не подходит, т. к. у тРНК 4 стебля). При Р=10 - 2 структуры, 3 стебля и в той и в другой. При Р=15 - 4 структуры, 3-ая содержит 4 стебля, остальные - по 3 стебля. При Р=20 были найдены те же 4 структуры, что и при Р=15, но к ним добавилась 5-ая, содержащая 5 стеблей и имеющая строение, нетипичное для тРНК. Дальнейшее увеличение Р-параметра я сочла ненужным, нам подходит структура №3, полученная при Р=15. Она и приведена на рис. 1.
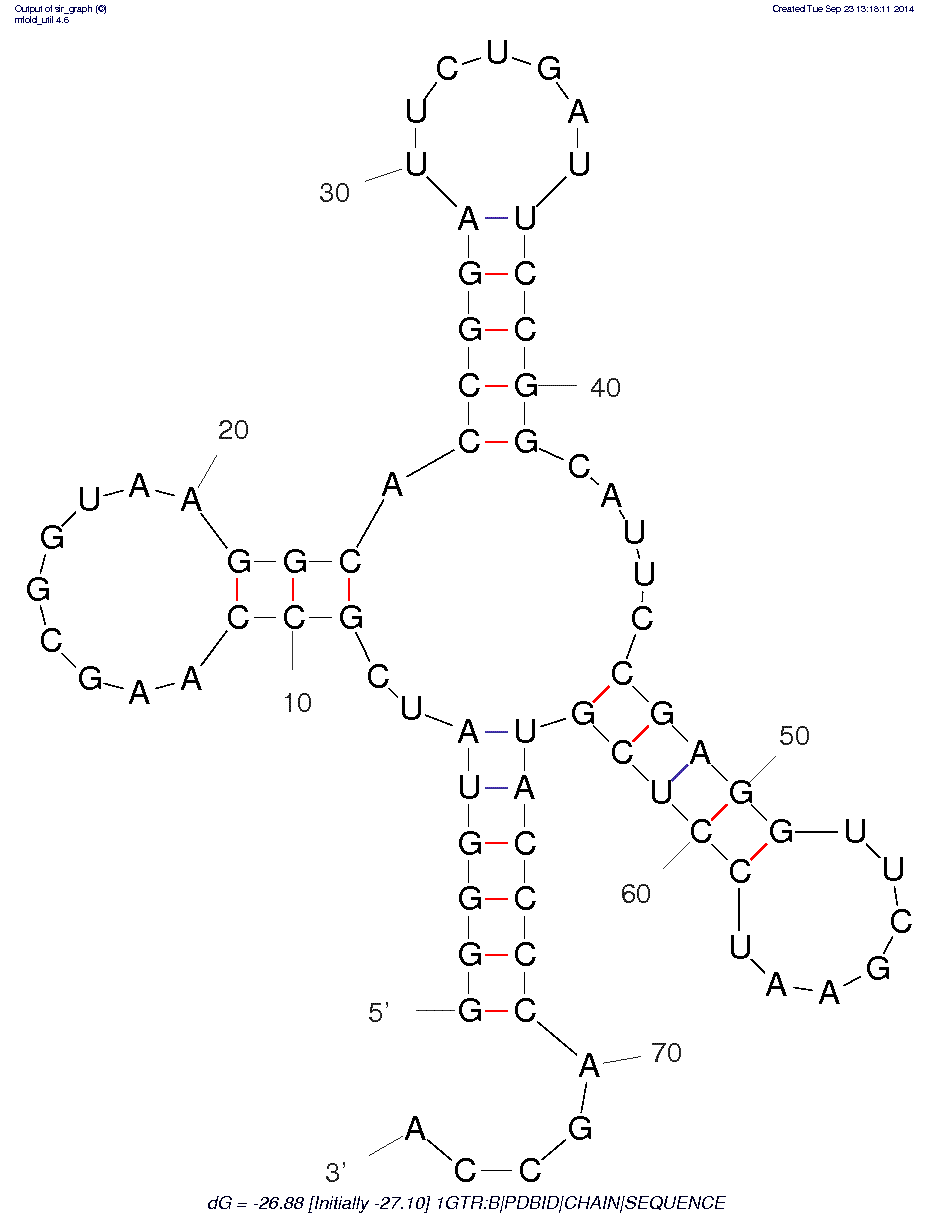
Рис. 1. Предсказанная вторичная структура тРНК из файла 1GTR.pdb. Изображение получено с помощью программы mfold.
Таблица 1. Сравнение кристаллической структуры тРНК 1GTR c предсказанными структурами этой тРНК в программах einverted и mfold
| Участок структуры | Позиции в структуре (по результатам find_pair) | Количество пар по результатам find_pair | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера (с помощью mfold) |
| Акцепторный стебель | 5'-3-7-2' и комплементарный участок 5'-66-70-3' | 5 пар | предсказано 5 пар | предсказано 6 пар |
| D-стебель | 5'-11-12-3' и комплементарный участок 5'-23-24-3' | 2 пары | - | предсказано 3 пары |
| T-стебель | 5'-50-53-3' и комплементарный участок 5'-61-64-3' | 4 пары | - | предсказано 5 пар |
| Антикодоновый стебель | 5'-38-44-3' и комплементарный участок 5'-26-32-3' | 7 пар | - | предсказано 5 пар |
| Общее число канонических пар нуклеотидов | 21 | 5 | 19 |
Поиск ДНК-белковых контактов в заданной структуре
Поиск ДНК-белковых контактов производился в помощью программы Jmol. Для этого сначала были определены следующие множества атомов:
- полярные атомы дезоксирибозы
- неполярные атомы дезоксирибозы
- полярные атомы остатков форфорной кислоты
- неполярные атомы остатков форфорной кислоты
- полярные атомы остатков азотистых оснований со стороны большой бороздки
- полярные атомы остатков азотистых оснований со стороны большой бороздки
- полярные атомы остатков азотистых оснований со стороны малой бороздки
- полярные атомы остатков азотистых оснований со стороны малой бороздки
- полярные атомы белка
- неполярные атомы белка
Таблица 2. Различные ДНК-белковые контакты в комплексе 1RH6.pdb
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 4 | 22 | 26 |
| остатками фосфорной кислоты | 11 | 17 | 28 |
| остатками азотистых оснований со стороны большой бороздки | 3 | 5 | 8 |
| остатками азотистых оснований со стороны малой бороздки | 1 | 0 | 1 |
С помощью программы nucplot была получена схема ДНК-белковых контактов. На вход программе подавался файл в старом формате PDB - 1RH6_old.pdb. Полученное изображение приведено на рис. 2. Исходный файл *.ps можно скачать здесь.
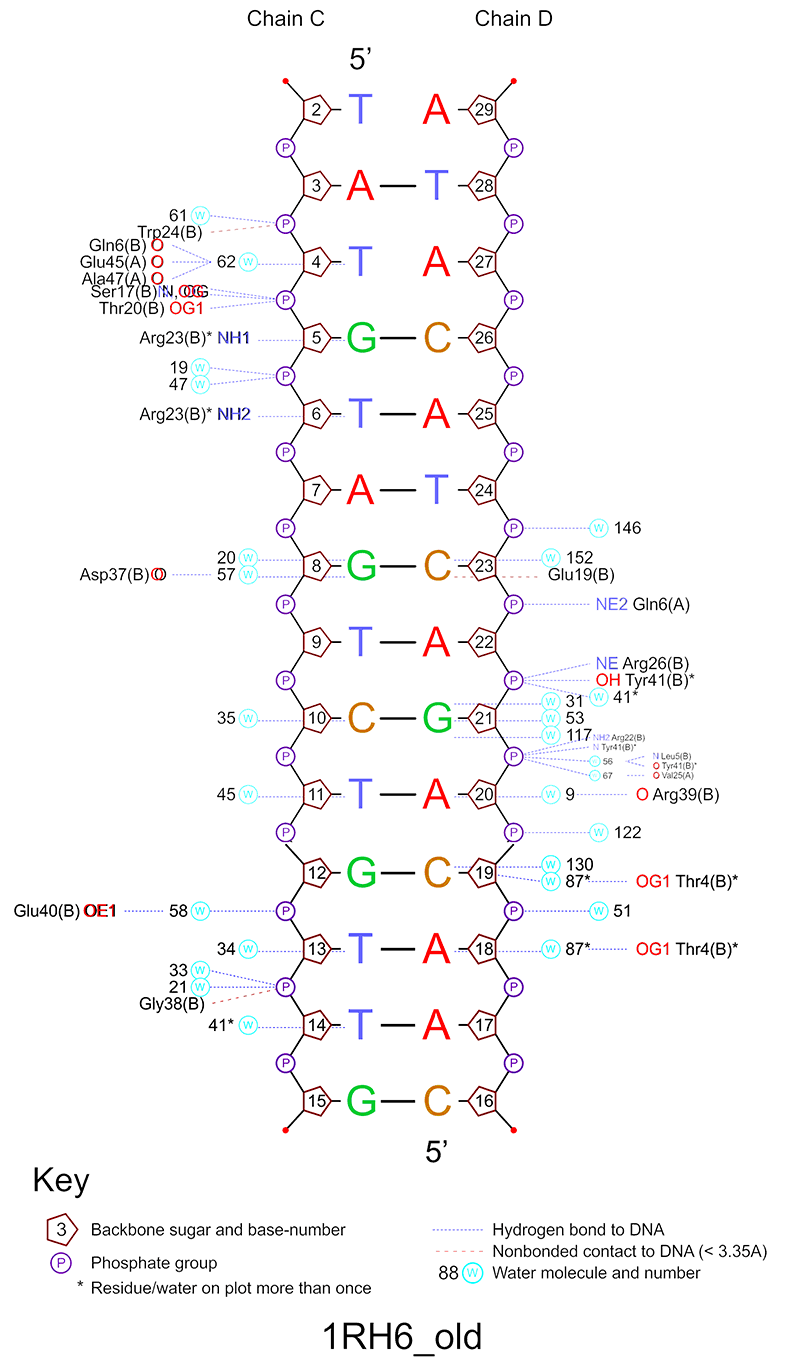
Рис. 2. Схема ДНК-белковых контактов структуры 1RH6. Изображение получено с помощью программы nucplot.
Аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК - Arg23 (2 контакта с ДНК). Thr4 также имеет 2 контакта с ДНК, но это контакты через молекулу воды. Наиболее важным для распознавания последовательности ДНК, по моему мнению, также будет Arg23, т. к. у него самое большое количество контактов с азотистыми основаниями ДНК. С помощью программы Jmol было создано изображение контактов Arg23 с ДНК. Оно приведено на рис. 3.
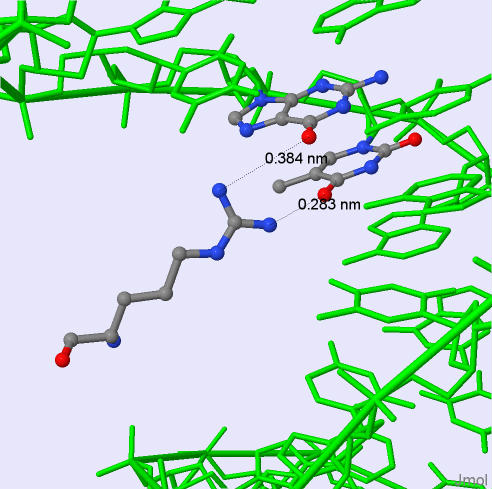
Рис. 3. Контакт Arg23 с ДНК из структуры 1RH6. Аминокислотный остаток Arg23 и связанные с ним азотистые остования (5-ый гуанин и 6-ой тимин) показаны в проволочной и шариковой модели, окраска по типу атомов. Остальная часть молекулы ДНК показана в проволочной модели и выделена зелёным цветом. Изображение получено с помощью программы Jmol.
© Shvetsova Ekaterina, FBB MSU, 2013
Дата последнего изменения: 07.12.2016