Задание1.
С помощью программ samtools и bcftools получить список однонуклеотидных
полиморфизмов (SNP) и инделей (то есть делеций и инсерций) для ридов, картированных на геномы хлоропласта и митохондрии из предыдущего задания.
Использованная команды:
samtools mpileup -u -g -f genomes.fasta out_sorted.bam > genomes.bcf
Чтобы получить список SNP и инделей, используем bcftools. bcftools view -vcg genomes.bcf > genomes.vcf Результат - файл со списком genomes.vcf. Число строк, содержащих 'DP=' соответствует числу SNP, а содержащих 'INDEL;' - числу инделей.
grep 'DP=' genomes.vcf | wc -l
grep 'INDEL;' genomes.vcf | wc -l
Количество инделей 295, а количество полиморфизмов 635.
Задание 2.
В этом задании нужно собрать геном хлоропласта и митохондрии на основе картированных ридов.
Характеристика N50 показывает длину контига, при которой 50% гипотетической
длины последовательности покрываются контигами длины, равной значению N50.
Нужно было перебрать различные длины k-меров, чтобы N50 оказался максимальным.
При длине k-мера = 25 N50 = 130, и это максимум среди тех длин, которые я перебрала.
velveth velveth_dir_31 31 -fastq f1.fastq
velvetg velveth_dir_31 -cov_cutoff auto
После этого я сделала локальный бласт контигов.
makeblastdb -in sequence.fasta -dbtype nucl
blastn -task blastn -query contigs.fa -db genomes.fasta -outfmt 7
-num_alignments 1 -out alignment.fa
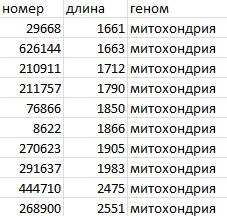
А потом с помощью Exel отобрала 10 самых длинных контигов в таблицу: