Комплексы ДНК-белок
1. Предсказание вторичной структуры заданной тРНК
1.1 Предсказание вторичной структуры тРНК путем поиска инвертированных повторов
Для структуры тРНК из прошлого практикума с помощью программы einverted были получены инвертированные участки в нуклеотидных последовательностях, которые могут образовывать стебли. При параметрах по умолчанию программа не давала результатов. Поэтому параметры были изменены на следующие:
lidia@kodomo:~/public_html/term3/block1/pr3$ einverted rcsb_pdb_1N77.fasta
Find inverted repeats in nucleotide sequences
Gap penalty [12]: 12
Minimum score threshold [50]: 0
Match score [3]: 20
Mismatch score [-4]: -4
Sanger Centre program inverted output file [emboss_001.inv]: 1n77.inv
File for sequence of regions of inverted repeats. [emboss_001.fasta]: 1n77.fasta
Вывод программы представлен ниже:
EMBOSS_001: Score 280: 22/28 ( 78%) matches, 13 gaps
1 ggccccatcgtctagcgg-ttaggacgcggccc-tctcaag 39
| ||||| | | || | | || | ||| | || |
71 ctggggt--c----ccccttagcttg-g--gggcaaag--c 42
Красным цветом обозначен стебель, совпадающий с предсказанием по алгоритму Зукера и с предсказанием с помощью find_pair. Видимо, программа einverted складывает последовательность примерно пополам, поэтому понятно, что боковые стебли в таком случае не будут обнаружены.
1.2 Предсказание вторичной структуры тРНК по алгоритму Зукера
Также вторичная структура тРНК была предсказана с помощью алгоритма Зукера, реализуемого программой RNAfold из пакета Viena Rna Package. Для этого использовалась веб-версия программы. На рисунке 1 показано графическое представление предсказания.
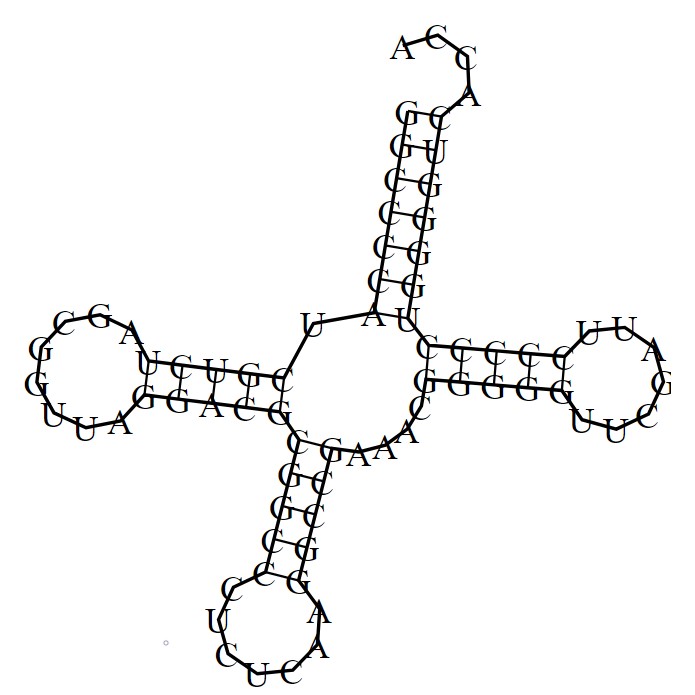
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
|---|---|---|---|
| Акцепторный стебель | 5'-503-507-3 3'-570-566-5' Всего 5 пар |
5 пар | 7 пар |
| D-стебель | 5'-510-512-3' 3'-525-523-5' Всего 3 пары |
0 пар | 5 пар |
| T-стебель | 5'-549-553-3' 3'-565-561-5' Всего 5 пар |
0 пар | 5 пар |
| Антикодоновый стебель | 5'-539-543-3' 3'-531-527-5' Всего 5 пар |
0 пар | 5 пар |
| Общее число канонических пар нуклеотидов | 18 | 22 (5 из них верные) | 22 |
2. Поиск ДНК-белковых контактов в заданной структуре (1A02 pdb-код)
2.1. Множества атомов в JMol
В данной части практикума в Jmol надо было задать следующие множества:
- Множество атомов кислорода 2'-дезоксирибозы (set1).
- Множество атомов кислорода в остатке фосфорной кислоты (set2).
- Множество атомов азота в азотистых основаниях (set3).
Скрипт с задаными множествами находится здесь.
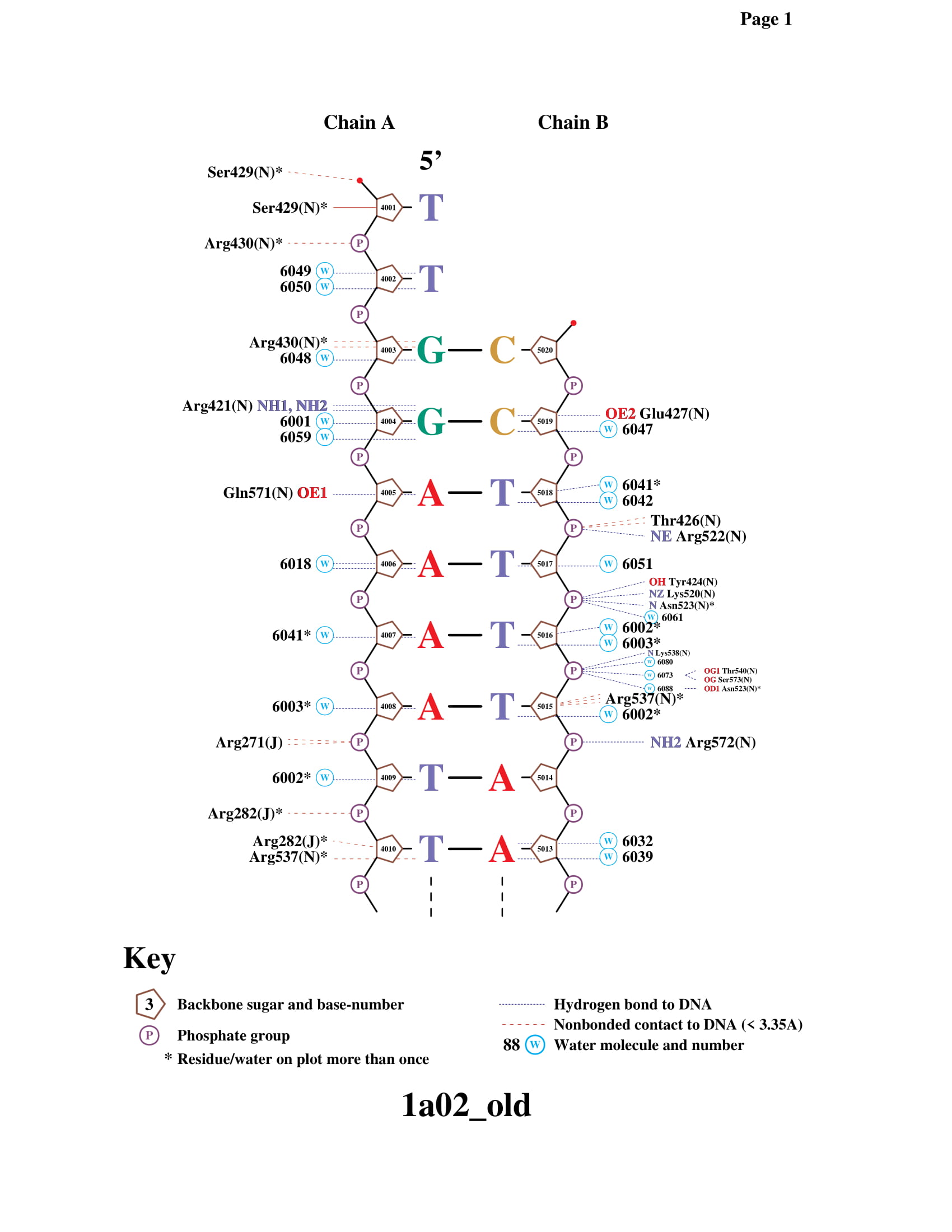
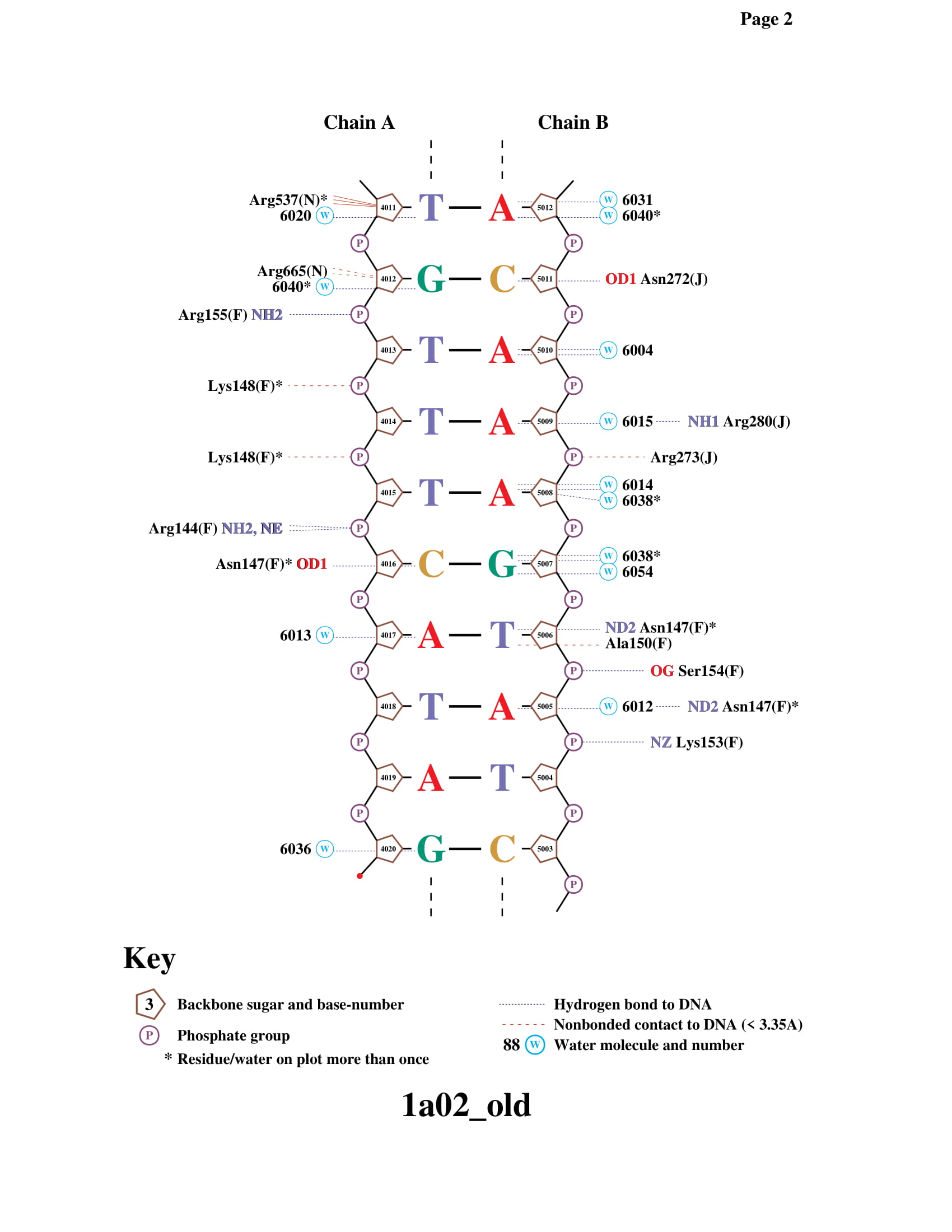
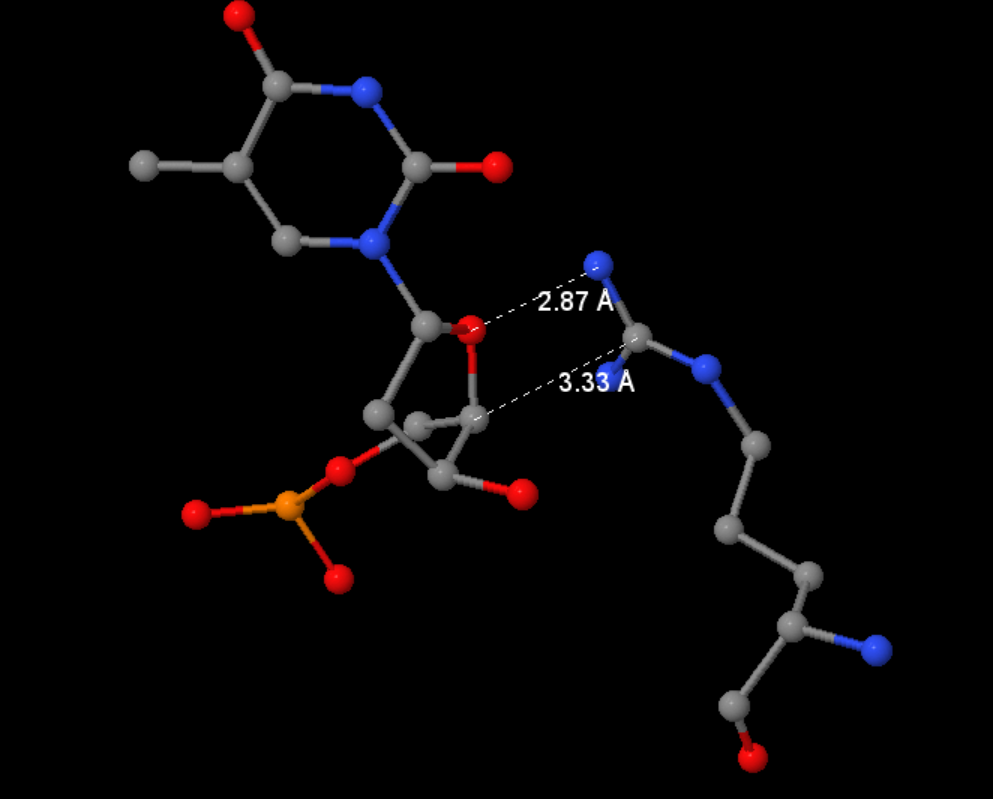
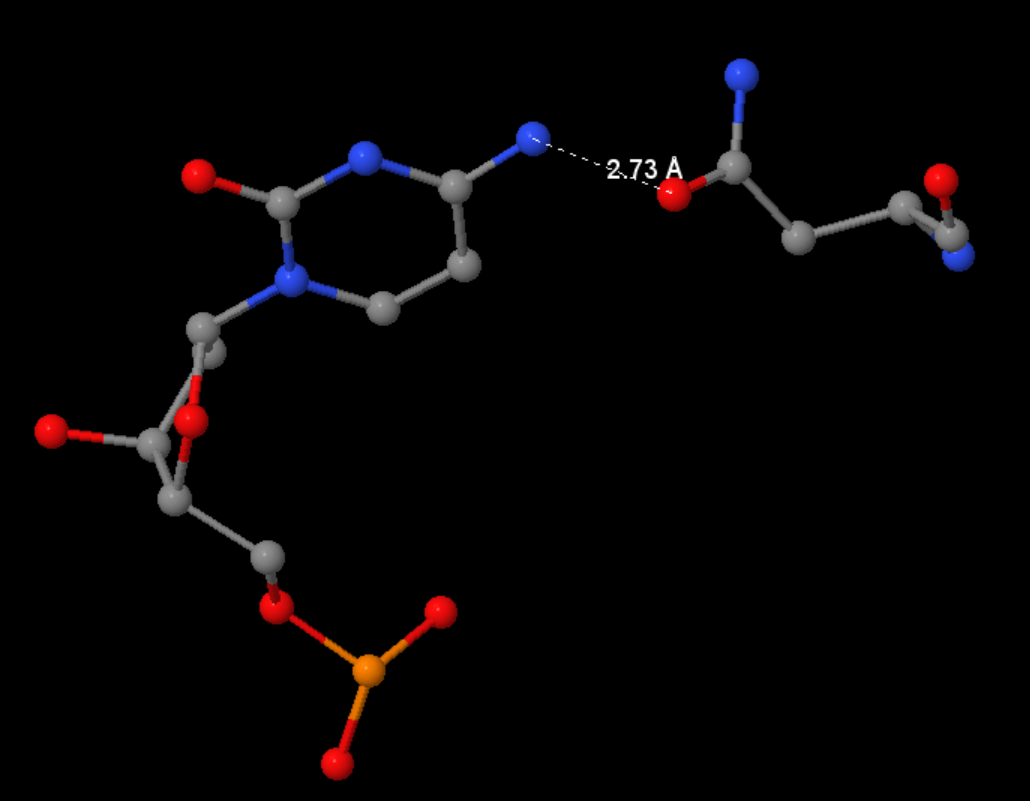
2.2. Описание ДНК-белковых контактов в заданной структуре
С помощью следующего скрипта были найдены ДНК-белковые контакты: полярный контакт - ситуация, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5Å. Аналогично, неполярный контакт - пара неполярных атомов на расстоянии меньше 4.5Å.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
|---|---|---|---|
| остатками 2'-дезоксирибозы | 4 | 10 | 14 |
| остатками фосфорной кислоты | 13 | 6 | 19 |
| остатками азотистых оснований со стороны большой бороздки | 7 | 12 | 19 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 1 | 1 |
2.3. Получение схемы ДНК-белковых контактов с помощью программы nucplot
Полный файл можно найти тут. Часть вывода представлена на рисунках 2 и 3 ниже.