Сравнение матриц аминокислотных замен
Матрицы типа BLOSUM62 и типа PHAT отличаются, в первую очередь, исходными данными, поступающими для обработки. Матрица BLOSUM62 ориентрована на белки цитоплазмы, тогда как при составлении матрицы типа PHAT использовались последовательности мембранных белков. Это было сделано из-за того, что у трансмембранных участков белков сильно изменены частоты аминокислот (меньше заряженных, больше неполярных). В связи с этим использование матрицы BLOSUM приводит к ошибочному пониманию эволюции этих участков.Одним из примеров матрицы, основанной на данных о последовательностях трансмембранных участков белков, является матрица PHAT (predicted hydrophobic and transmembrane regions, A Transmembrane-Specific Substitution Matrix), основанная на гидрофобных и трансмембранных доменах в последовательностях белков из базы данных Blocks. .
Матрица весов замен BLOSUM62 имеет порог кластеризации 62%. Кластеризация нужна для учета очень близких, практически идентичных последовательностей, слишком родственных друг другу. Замены в них нельзя считать случайными, поэтому близкие последовательности кластеризуют, то есть объединяют; их считают за одну последовательность. Соответственно вклад идентичных последовательностей уменьшается.
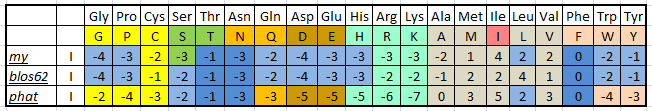 |
| Рис.1. Веса замен по различным матрицам для изолейцина. Разными цветами выделены аминокислоты, сходные по свойствам. Оттенками синего выделены одинаковые веса разных матриц. |
Заметим, что матрица замен PHAT, ориентированная в основном на незаряженные аминокислотные последовательности, сильнее штрафует за замены алифатической неполярной аминокислоты изолейцина на заряженные или полярные. И дает больше баллов за замены изолейцина на другие неполярные аминокислоты. В том числе, замена остатка изолейцина на такой же прекрасно иллюстрирует эту тенденцию.
Данные по весам замен у матрицы, полученной мной и классической матрицей BLOSUM62, отличаются по некоторым аминокислотам. Это связано с тем, что матрица BLOSUM62 использовала базу данных последоватеьлностей 1992 года; с того момента BLOCKS неоднократно пополнялся; соответственно, частоты аминокислот слегка изменились, то и привело к некоторым разногласиям.
Сравнение выравниваний, полученных для коротких мутантов вручную и построенных классическими алгоритмами Нидлмана-Вунша и Смита-Ватермана
Алгоритмы, использующиеся при глобальном и локальном выравнивавниях, реализуются программами needle и water соответственно. Обе эти программы являются частью пакета EMBOSS.
Алгоритмы имеют сходные принципы, но отличаются деталями. Если в глобальном выравнивании мы ищем оптимальный способ выровнять всю последовательность со всей последовательностью, потеряв при этом как можно меньше очков (штрафов за гэпы), то при локальном подход другой: нам нужно найти только часть последовательности, которая будет сильно похожа на кусочек другой последовательности. Соответственно штрафы за гэпы при глобальном выравнивании существуют всегда; локальное выравнивание не обращает внимание на пропуски вне выровненного кусочка. Если там будет много гэпов -выравнивание просто "оборвется" на данном моменте, если же нет - этот участок будет всключен в выравнвание и подсчет штрафов за гэпы будет аналогичным таковому в алгоритме глобального выравнивания. Что касается цены за совпадение или несовпадение, баллы одинаковы для двух алгоритмов.
Алгоритм needle и water используют следующие параметры (для нескольких приведены значения по умолчанию, другие вводятся самотоятельно):
- gapopen (штраф за открытие гэпа): 10
- gapextend (штраф за продолжение гэпа): 0.5
- datafile (матрица весов замен): EBLOSUM62 для белков, EDNAFULL для нуклеиновых кислот (здесь и далее НК)
- endweight (штраф за "концевые" гэпы)
- endopen (штраф за открытие "концевого" гэпа): 10
- endextend (штраф за продолжение "концевого" гэпа): 0.5
 |
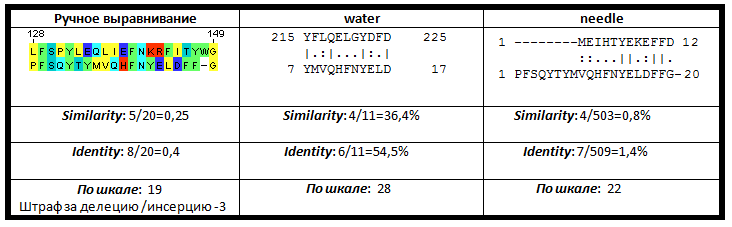
| Рис. 1. Сравниние выравниваний для мутанта 1 |
Ручное выравнивание по сравнению с работой алгоритмов needle и water в случае выравнивания первого мутанта оказалось самым качественным. Алгоритм для локального выравнивания последовательностей избавился от трех концевых аминокислот пептида, так как выравнивание их приводит к обязательному появлению гэпа, вес которого, при стоящих по умолчанию величинах штрафов за гэпы, оказыватеся меньше. Глобальное выравнивание последовательности подтверждает это.
Однако из-за высокой вероятности появления вставки или делеции внутри мутантного пептида позволяет нам не штрафовать за появление оных так сильно. Штрафы, используемые в алгоритмах по умолчанию, подразумевают естественную эволюцию белков, при которой эти вероятности гораздо меньше. Поэтому реальный вес выравнивания оказывается больше. Это же касается и весов выравниваний для мутантов 2 и 3.
 |
| Рис. 2. Сравниние выравниваний для мутанта 2 |
Для второго мутанта все три подхода дали абсолютно разные выравнивания. Заметим, что алгоритм water вновь "обрезал" пептид, оставив только хорошо выравнивающуюся часть. Needle неплохо выровнил последовательность, но выравнивание неправильное, потому что пептид является частью исходного мутировавшего пептида, поэтому предположение needle о вставке целых 8 аминокислот впереди пептида неверно.
 |
| Рис. 3. Сравниние выравниваний для мутанта 3 |
Лишь для выравниваний третьего мутанта мы получили сходящиеся результаты. Это связано главным образом с отсутствием гэпов в выравниваниях.