1. Построение дерева по нуклеотидным последовательностям.
Построить филогенетическое дерево тех же бактерий, что в предыдущих заданиях, используя последовательности РНК малой субъединицы рибосомы (16S rRNA).
В Uniprot найдем любые белки бактерий. Найдем ссылки на полные геномы записей EMBL. Там найдем FT с ключом "rRNA". Полученные данные приведены в таблице:
| Название | Мнемоника | AC записи EMBL | Координаты РНК | Цепь |
| Bacillus anthracis | BACAN | AE016879 | 29129..30635 | Прямая |
| Clostridium botulinum | CLOB1 | CP000726 | 9282..10783 | Прямая |
| Enterococcus faecalis | ENTFA | AE016830 | 1018187..1019708 | Прямая |
| Geobacillus kaustophilus | GEOKA | BA000043 | 30790..32343 | Прямая |
| Lactobacillus delbrueckii | LACDA | CR954253 | 45160..46720 | Прямая |
| Listeria monocytogenes | LISMO | AL591981 | 99187..100732 | Обратная |
| Staphylococcus epidermidis | STAES | AE015929 | 1722288..1723841 | Обратная |
| Streptococcus pneumoniae | STRPN | FM211187 | 16806..18218 | Прямая |
Вырежем нужные участки из записи EMBL командой seqret:
seqret embl:xxxxxxxx -sask
Поместим последовательности в один fasta-файл all.fasta. Отредактировали названия последовательностей (чтобы было удобнее).
Создадим выравнивание отобранных белков. Для этого на kodomo запустим программу muscle с параметрами по умолчанию.
muscle -in all.fasta -out align.fastaВыравнивание в формате fasta
Дерево, построенное по выравниванию 16s рРНК из заданных бактерий программой MEGA (Neigbour joining tree)
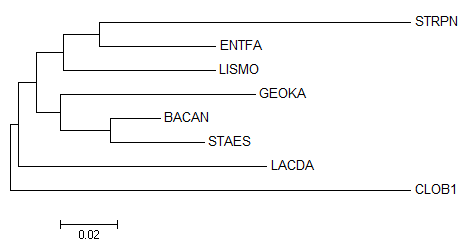
Правильное дерево
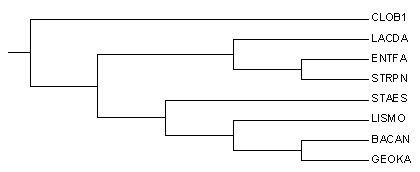
Эти деревья достаточно сильно различаются. На мой взгляд, это можно объяснить тем, что хотя рРНК довольно консервативна в близкородственных видах, все равно нуклеотидные последовательности более изменчивы, чем белки (из-за того, что аминокислоты кодируются триплетами, и не все нуклеотидные мутации влияют на замену аминокислоты).
2. Построение и анализ дерева, содержащего паралоги.
Чтобы найти гомологов в заданных организмах, воспользуемся файлом proteo.fasta на диске P, где лежат записи банка UNIPROT, относящиеся к бактериям, перечисленным в таблице к заданию 1. Проведем поиск программой blastp гомологов (с порогом на E-value 0,001) и отберем по мнемонике видов только те находки, которые относятся к отобранным ранее бактериям.
makeblastdb -in proteo.fasta -dbtype prot -out prot blastp -query clpx_bacsu.fasta -db prot -out gomolog.txt -evalue 0.001
На выдаче получили такой файл gomologs.txt
Нашлись такие гомологи из выбранных бактерий:
gomologs_ids.txt.Создали файл с последовательностями этих белков gomologs.fasta. Выровняли последовательности - файл align_gomologs.fasta.
muscle -in gomologs.fasta -out align_gomologs.fasta
Построили дерево методом Neighbor-Joining в программе MEGA
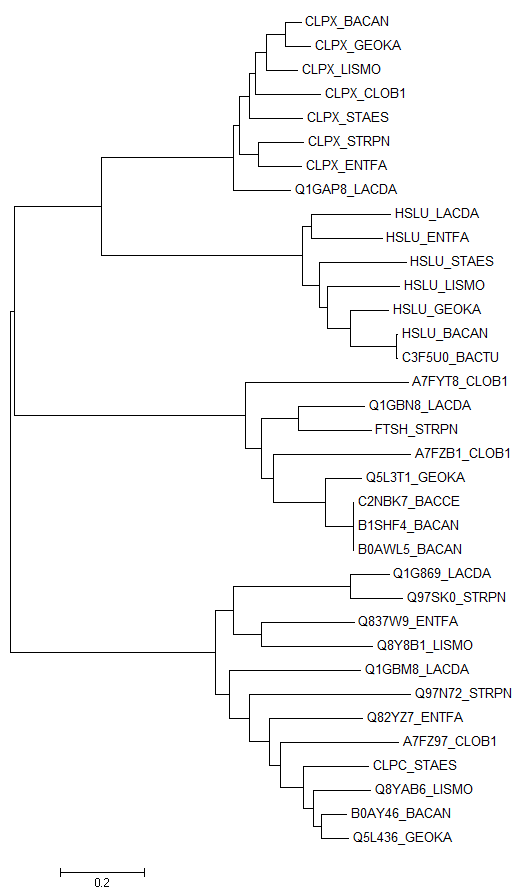
Если считать дерево верным, в нем можно выделить такие ортологи:
CLPX_BACAN - CLPX_GEOKA, CLPX_STRPN - CLPX_ENFTA, HSLU_LACDA - HSLU_ENTFA
Также можно выделить паралоги B1SHF4_BACAN - B0AWK5_BACAN.