d1. Определение вторичной структуры
Программа Stride
Структура белка 1VLI (с которой работала ранее) подходит для выполнения этого задания.
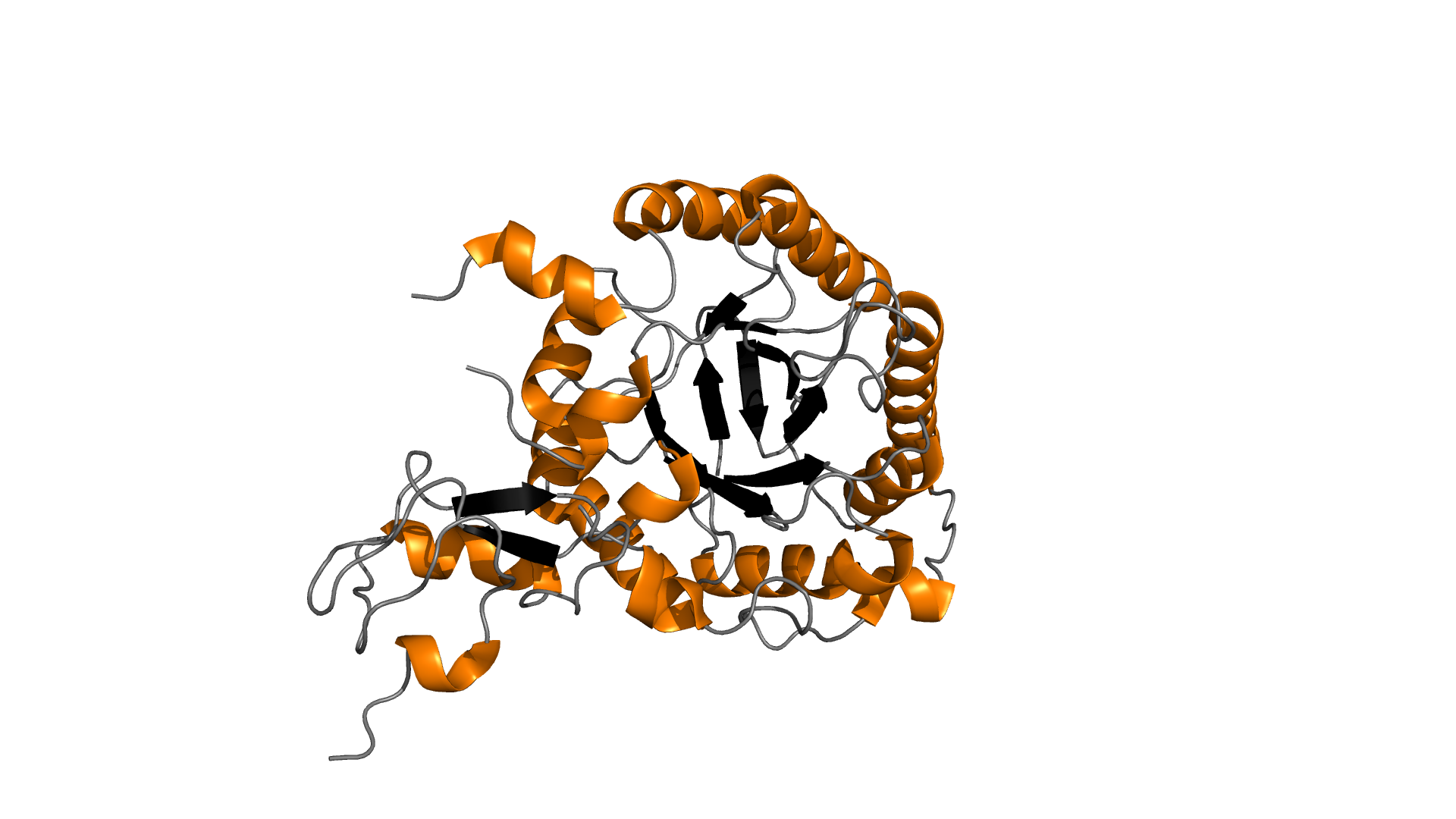
Для предсказания вторичной структуры использовался сервис Stride . Помимо текстовой выдачи , сервис позволяет также картировать вторичные структуры на последовательность белка.


| № элемента вторичной структуры | Аннотация в pdb-файле (номера остатков) | Результат Stride (номера остатков) |
| α-спираль | ||
| 1 | 30 – 45 | 31 – 44 |
| 2 | 55 – 60 | - |
| 3 | 76 – 81 | 77 – 82 |
| 4 | 82 – 84 | - |
| 5 | 90 – 101 | 91 – 101 |
| 6 | 111 – 120 | 112 – 120 |
| 7 | 135 - 144 | 136 – 144 |
| 8 | 157 - 170 | 158 – 170 |
| 9 | 219 - 228 | 219 - 228 |
| 10 | 253 - 274 | 254 - 273 |
| 11 | 280 - 285 | 281 - 284 |
| 12 | 339 - 345 | 340 - 344 |
| β-тяж | ||
| 1 | 3 - 6 | 3 - 6 |
| 2 | 9 - 12 | 9 - 12 |
| 3 | 18 - 24 | 18 - 24 |
| 4 | 48 - 51 | 48 - 51 |
| 5 | 104 - 106 | 104 - 107 |
| 6 | 126 - 128 | - |
| 7 | 149 - 152 | - |
| 8 | 176 - 181 | 177 - 181 |
| 9 | 208 - 213 | 208 - 213 |
| 10 | 232 - 236 | 232 - 236 |
| 11 | 305 - 308 | 305 - 308 |
| 12 | - | 316 - 318 |
| 13 | 323 - 326 | 323 - 326 |
| 14 | - | 347 - 349 |
| 15 | - | 357 - 358 |
| 310 спирали | ||
| 1 | 87 - 89 | 88 - 90 |
| 2 | 129 - 133 | 130 - 132 |
| 3 | 188 - 192 | 189 - 191 |
| 4 | 193 - 194 | 194 - 196 |
| 5 | 195 - 204 | 198 - 201 |
| 6 | 336 - 338 | 337 - 339 |
| 7 | 360 - 364 | 361 - 363 |
Помимо представленных в таблице вторичных структур, Stride выдает остатки для так называемых поворотов (Turns) и изолированных бета - мостиков. Для поворотов это остатки под номерами:
7 - 8, 13 - 17, 25 - 30, 133 - 135, 182 - 188, 202 - 207, 238 - 251, 309 - 322, 345 - 346. Изолированные бета-мосты из остатков 108, 127, 128, 151, 237, 312, 353. Примечательно, что остатки
126 - 128 помечены как бета-тяж, а 127 и 128 в Stride обозначились как изолированные бета-мосты.
В общем видно, что для бета-тяжей практически все границы совпадают в pdb-файле и Stride, а в альфа-спиралях часто встречается сдвиг на один аминокислотный остаток.
Также видим, что нашлись такие вторичные структуры, которые в одном из двух случаев отсутствуют. Часть из них сервис Stride приписал другим вторичным структурам, а для остальных случаев
напротив остатков, помеченных в pdb как вторичные структуры, в Stride стоит метка "Coil".
Сервис Sheep
Вместо сервиса Sheep (который не работал на момент выполнения задания) был использован сервис ProtOn , который также выполняет Sheep. Была построена карта бета-листа структуры 1VLI (использовались параметры по умолчанию), который представляет собой бета-баррель в структуре белка.

По карте видно, что выбранный бета-лист (а, точнее, бета-баррель) состоит из 8 тяжей.
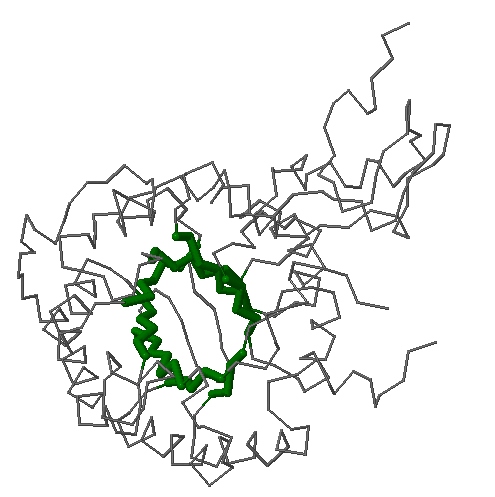
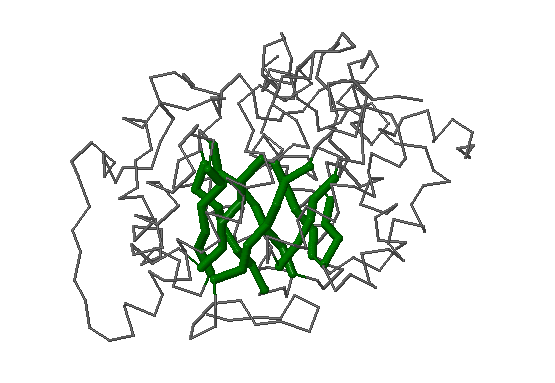
Один столбец карты соответствует одному хребту бета-листа. Был выбран 6 столбец (Thr-153, His-180, Phe-211, Ile-233, Ile-19, Asp-47).
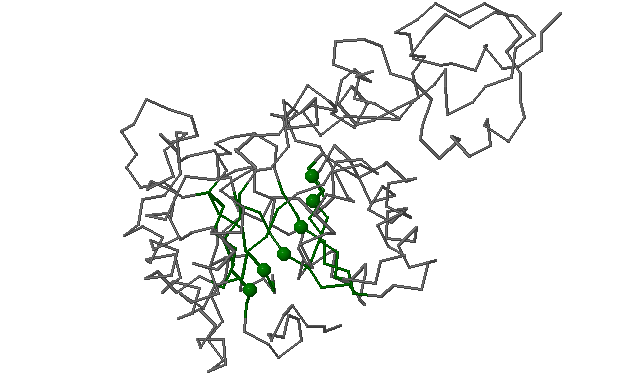
Выбранный бета-лист имеет почти одинаковое количество гидрофобных остатков с каждой стороны листа.
d2. Совмещение структур
Совмещение структуры 1VLI и структурных гомологов.С помощью сервиса PDBeFold были выбраны 4 структурных гомолога 1VLI: 1XUZ:A, 4IPI:A, 3G8R:B, 3NVT:B. Для пяти структур были загружены выравнивание последовательностей по совмещению структур ( множественное и парное ) и само совмещение структур.
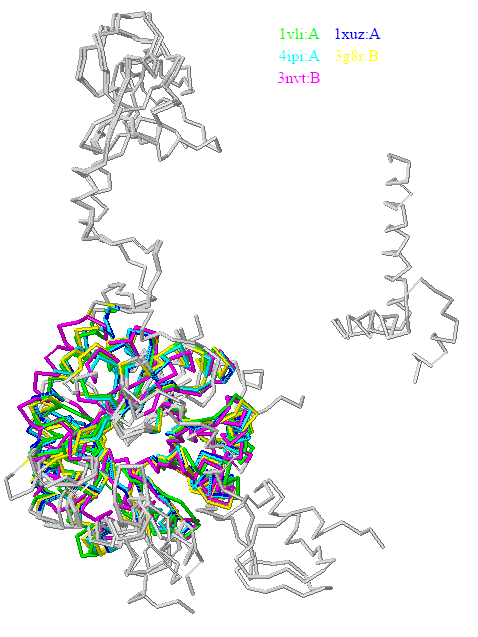
Также было произведено выравнивание программой JalView (с помощью Tcoffee со стандартными параметрами).

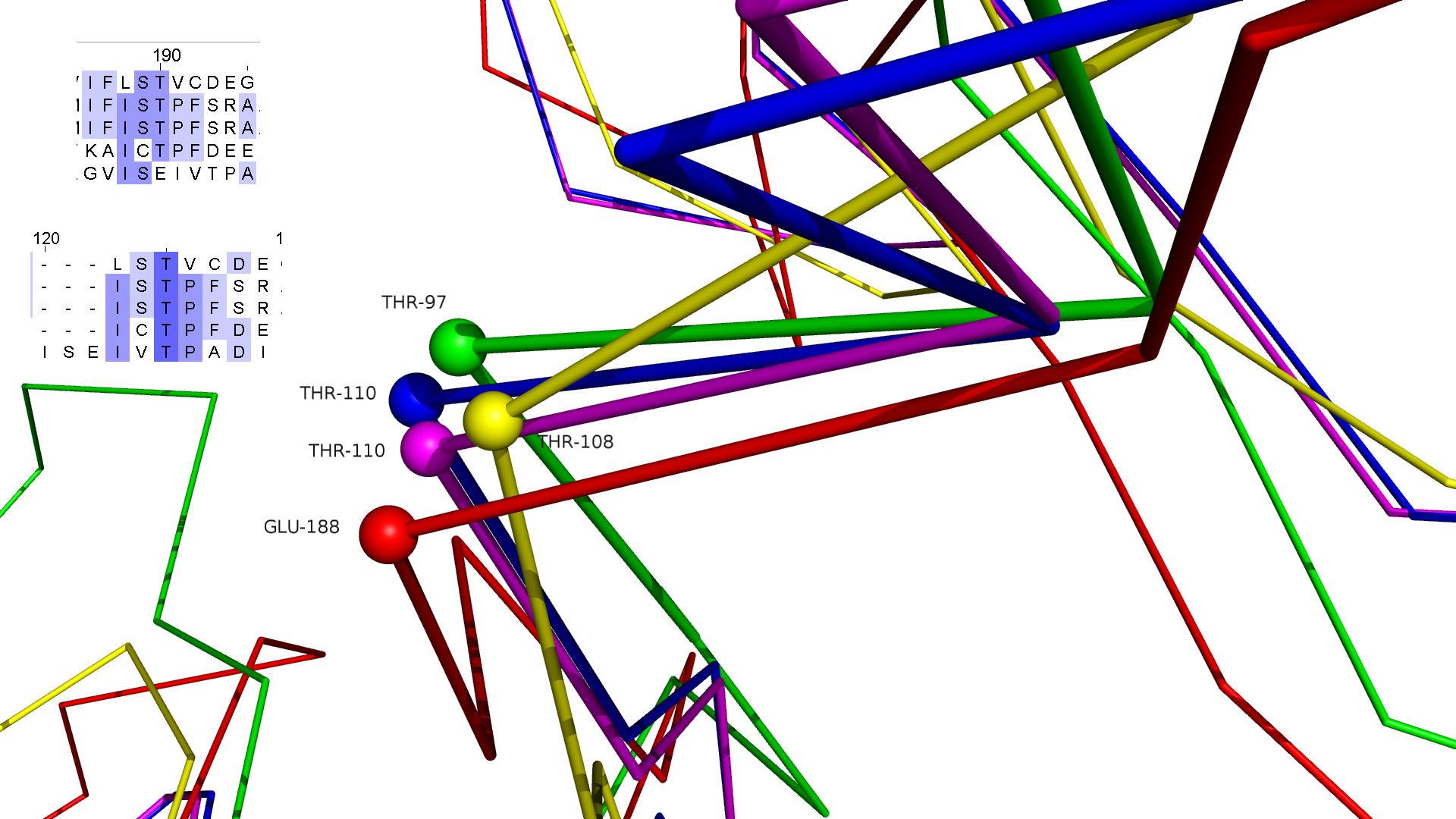
Виден ряд несоответствий в выравниваниях. Так, например, в выравнивании последовательностей по структурам есть консервативная позиция треонинов (в 1vli это 98 остаток), в то время как во множественном выравнивании последовательностей треонин 162 из структуры 3nvt:B не выровнялся с остальными.
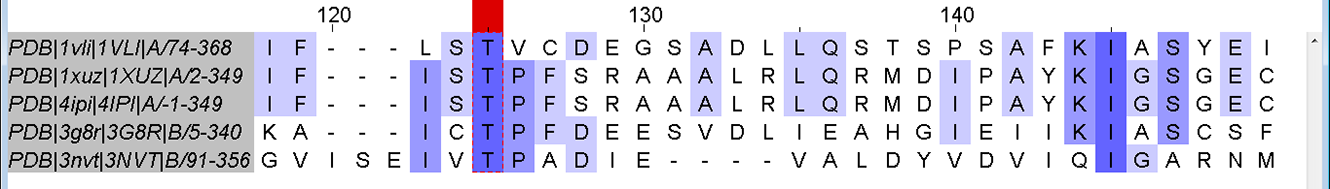

Ниже приведено изображение остатков, выровненных в случае выравнивания по структурам. Вероятно, выравнивание по структурам более правильное.
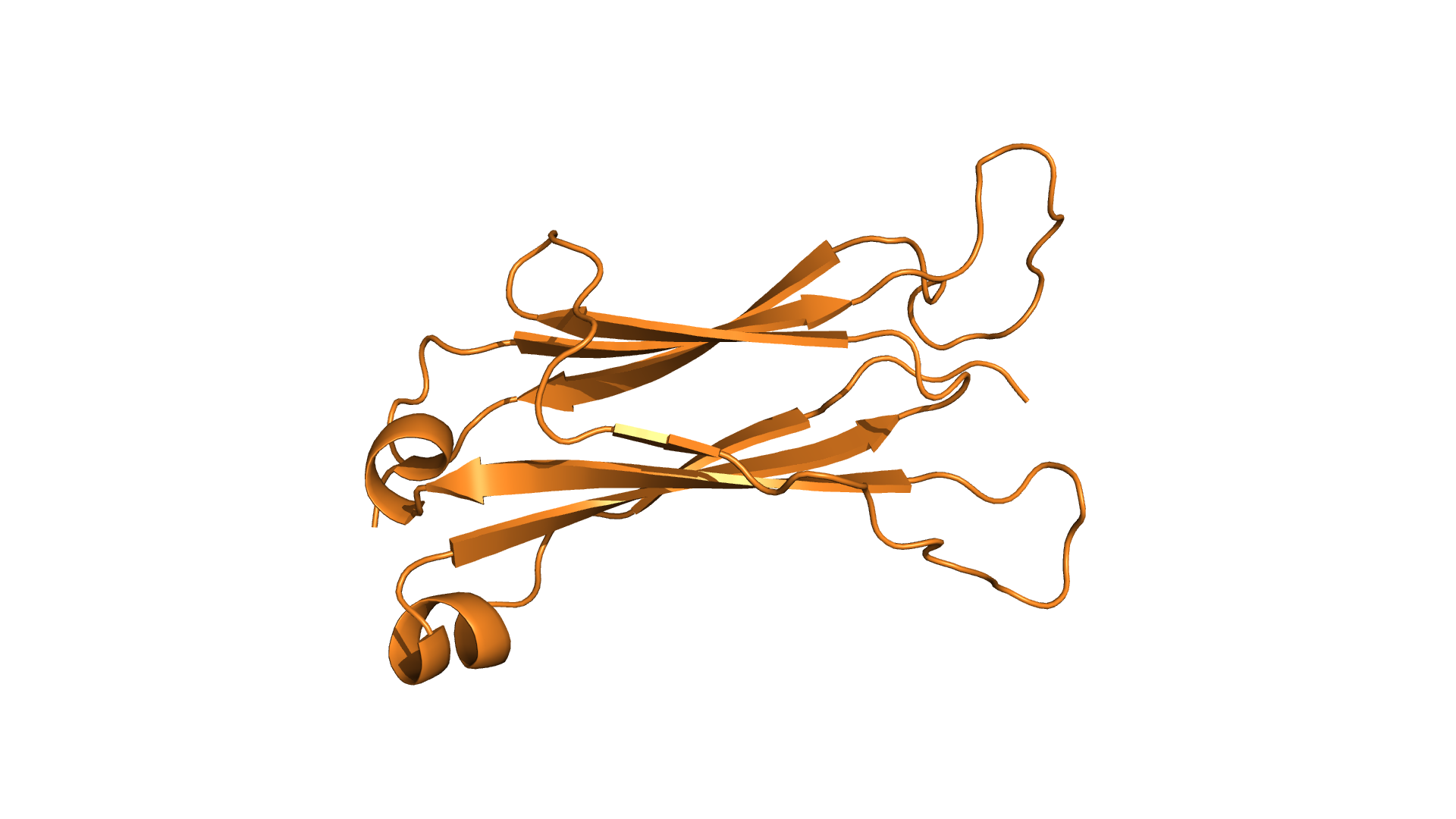
Совмещение по заданному выравниванию Были выбраны структуры константного домена человеческого Т-клеточного рецептора из цепи альфа (1oga, region d: 118-202) и из цепи бета (1oga, region e: 119 - 245). Формат .pdb: alpha.pdb и beta.pdb .
С помощью сервиса Sheep были получены карты бета-листов доменов
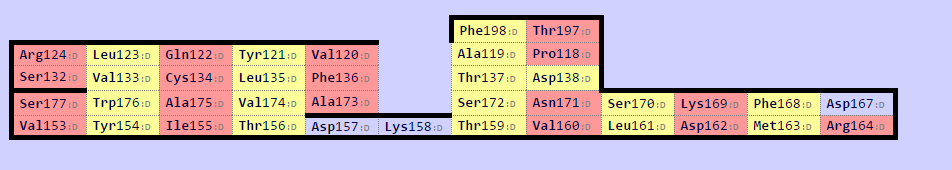
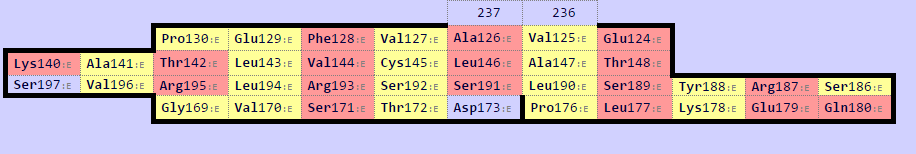
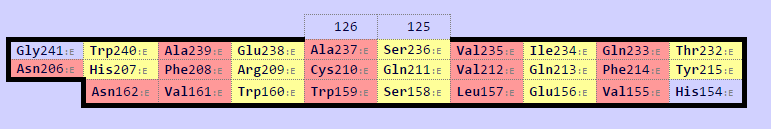
Карты map0 обоих доменов в одинаковой ориентации, и соответствуют друг другу. Можно построить выравнивание этих двух доменов. Консервативные остатки цистеина в них задают выравнивание центрального тяжа. Остатки, спаренные с консервативным цистеином, задают выравнивание "соседних тяжей". Команда для совмещения этих листов в PyMol (с использованием информации о выровненных остатках):
select s1, alpha & i. 122 & n. ca or alpha & i. 133 & n. ca or alpha & i. 134 & n. ca or alpha & i. 135 & n. ca or alpha & i. 155 & n. ca or alpha & i. 175 & n. ca select s2, beta & i. 127 & n. ca or beta & i. 144 & n. ca or beta & i. 145 & n. ca or beta & i. 146 & n. ca or beta & i. 172 & n. ca or eta & i. 192 & n. ca pair_fit s1, s2
Совмещение сохранено в соответствующем файле
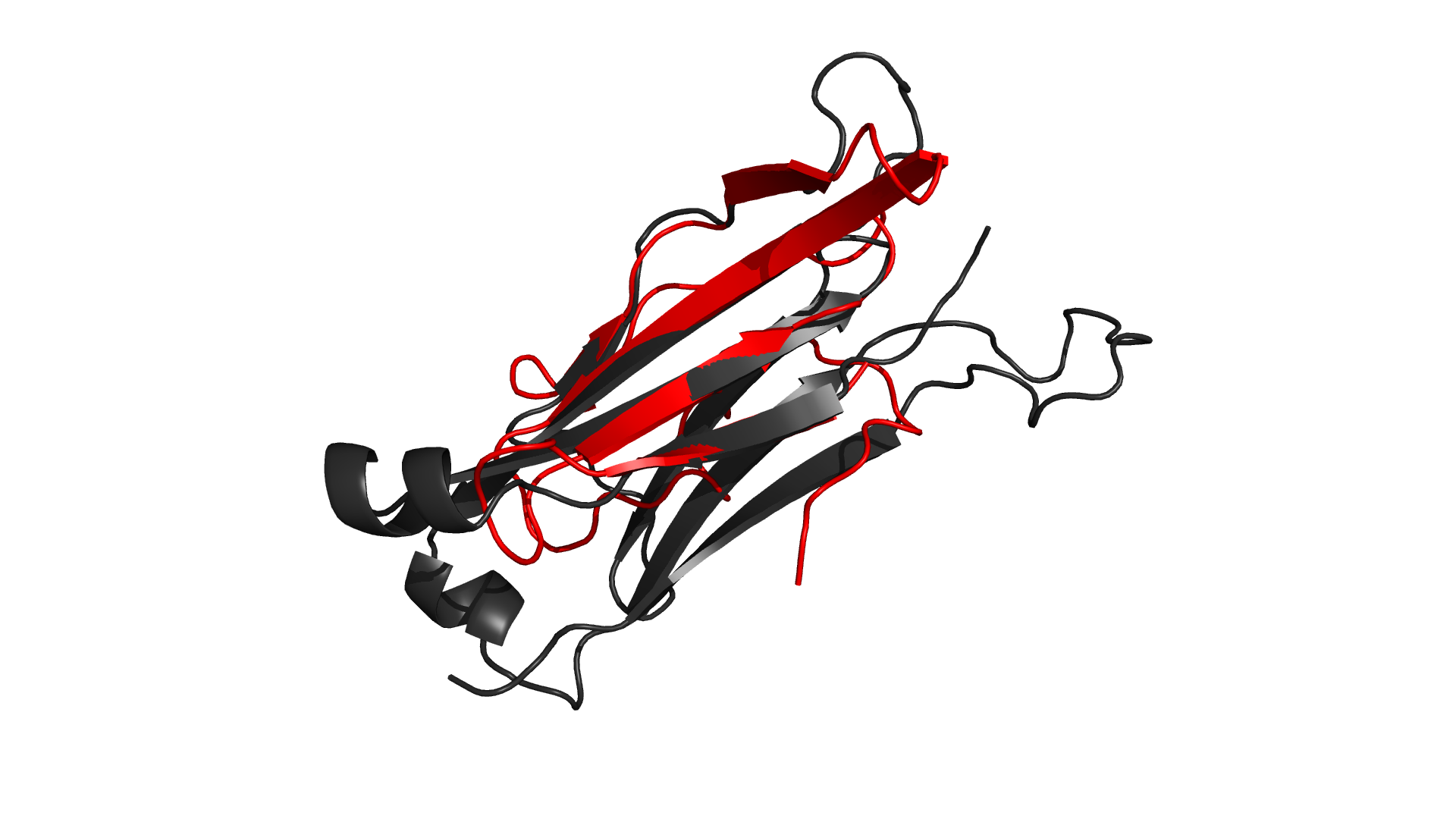
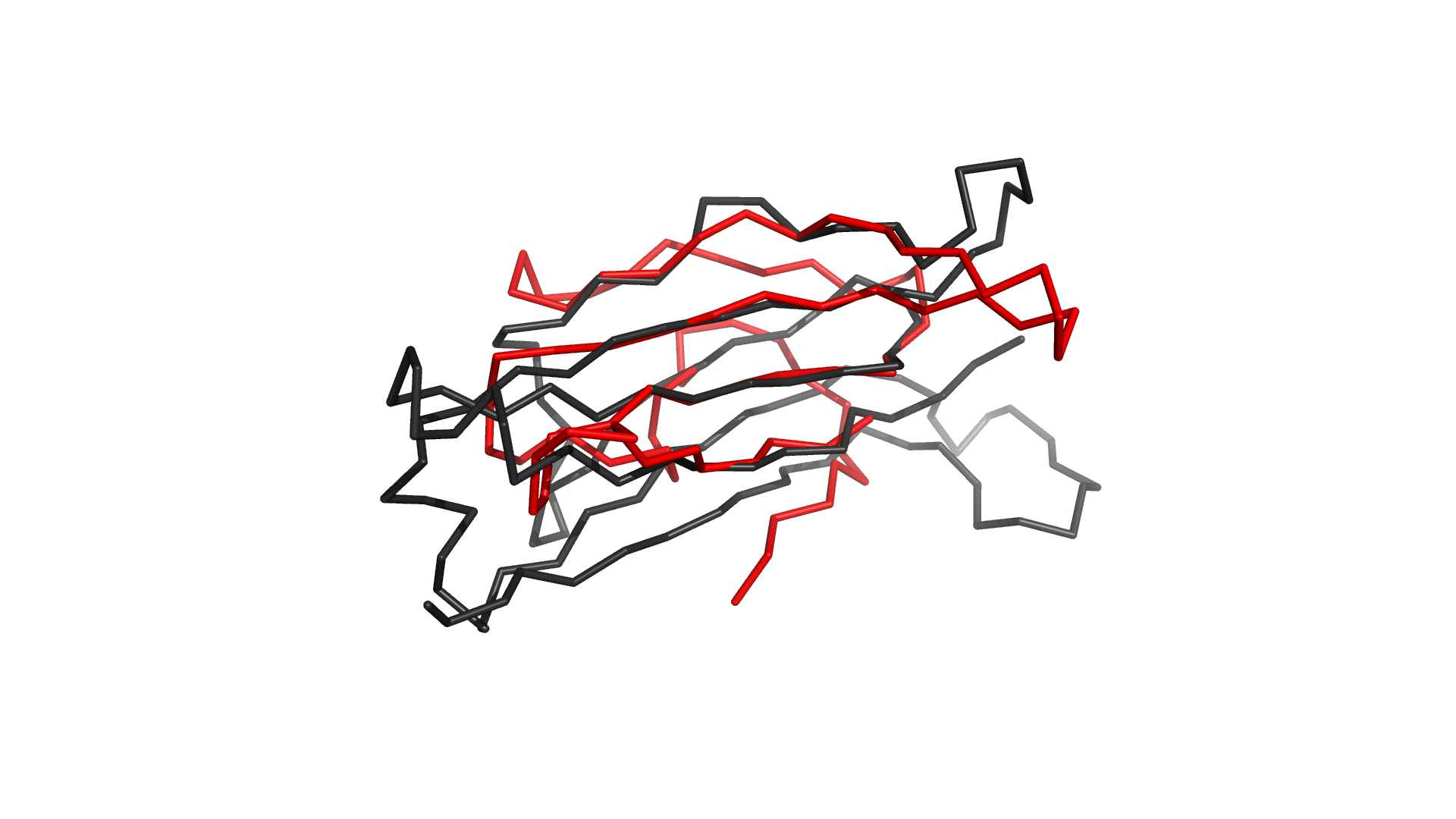
По совмещению видно, что ход полипептидной цепи в пространстве совпадает, то есть топологии сходны. Также видно соответствие всех петель в структурах.
d3. Нахождение гидрофобных кластеров
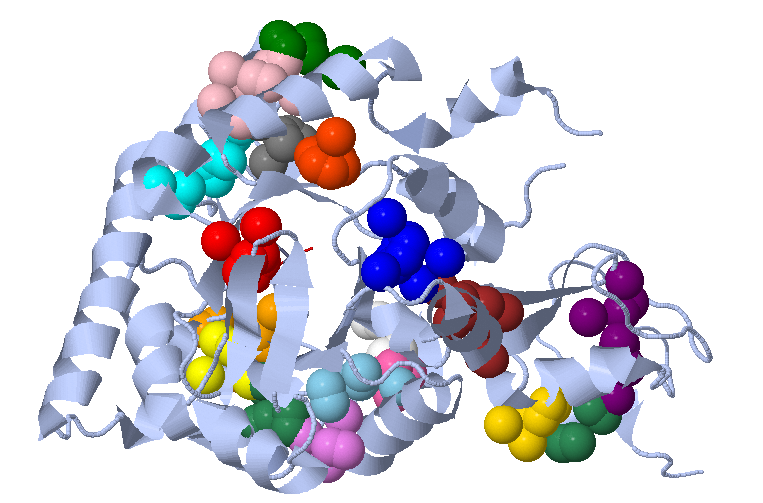
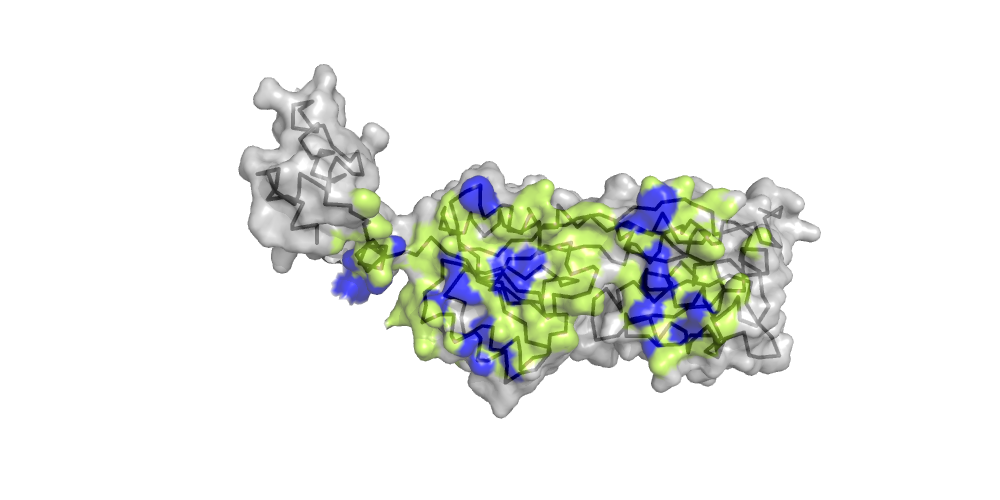
С помощью сервиса CluD в структуре 1VLI был происзведен поиск гидрофобных кластеров. Со значениями 5.3 для порога расстояния и 10 для размера кластера было найдено 3 кластера, изображенных ниже.
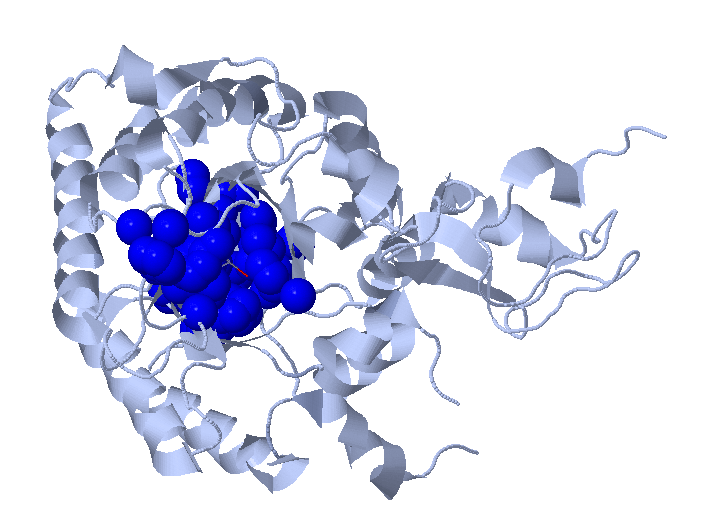
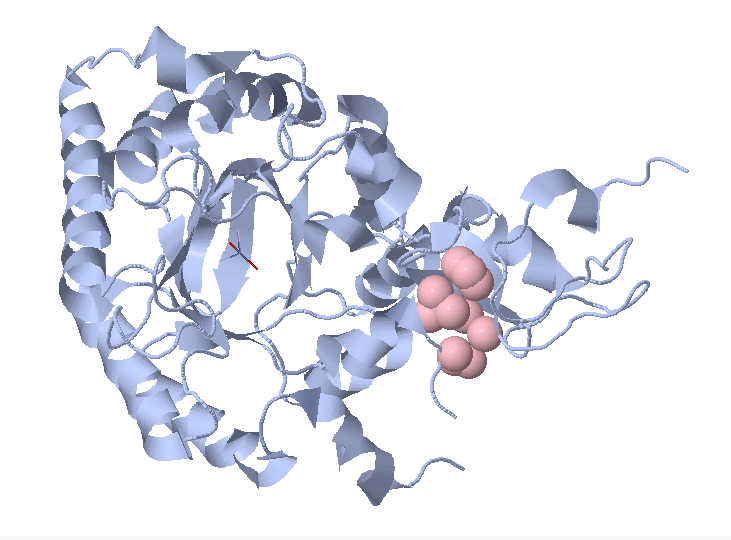
Первый кластер соответствует бета-баррелю структуры, в котором находится активный центр белка и где происходит связывание субстрата. Остальные же два кластера в точности соответствуют
доменам из Pfam этого белка - домену NeuB , который присущ всем синтазам N-ацетилнейраминовой кислоты прокариот, и домену SAF , который присущ многим разным белкам, а функция его расплывчато описывается как "метаболизм".
Также пробовала брать другие пороги расстояния и размера кластера.
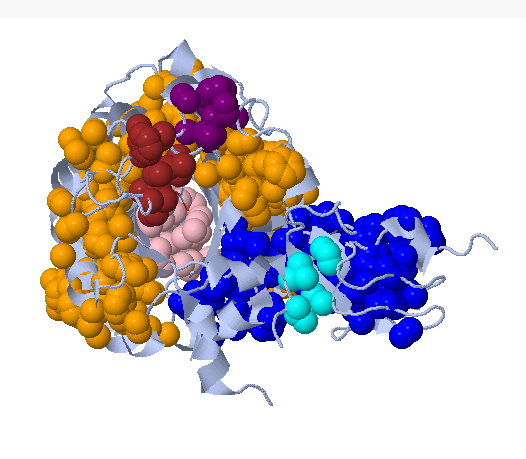
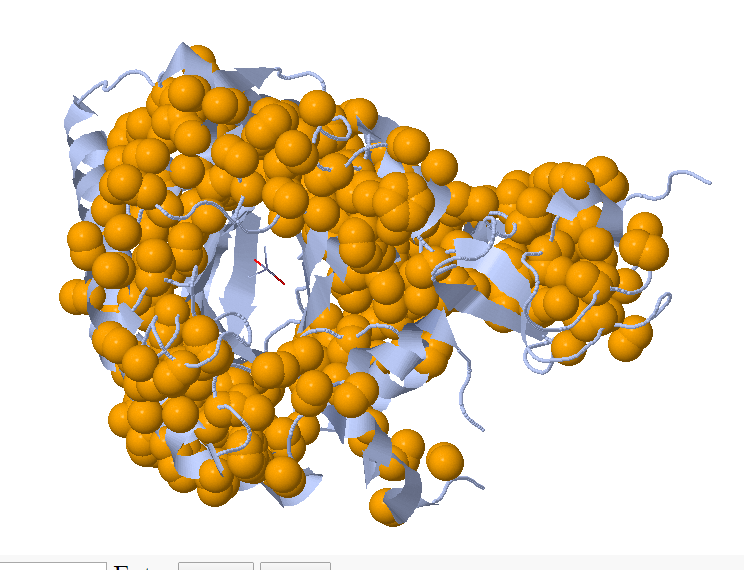
При пороге на расстояния 4.7 и пороге на размер кластера 10 видим, что кластеров находится больше, однако, идея осталась той же - кластерами покрыт весь белок. Но при таком распределении кластеров довольно сложно интерпретировать результат.
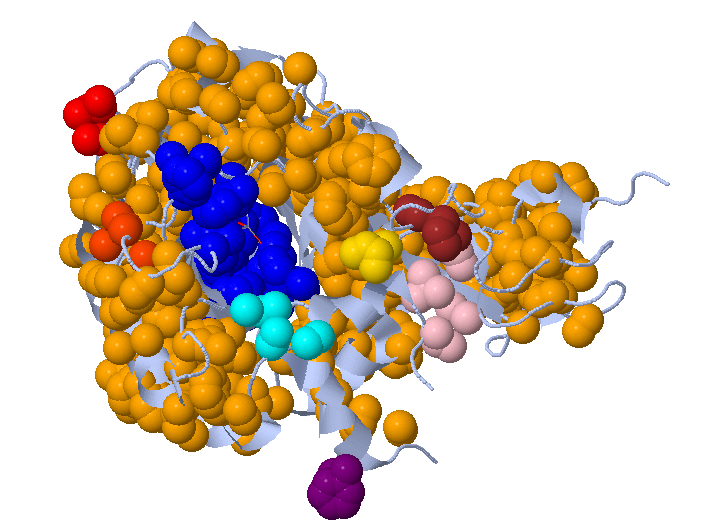
При порогах 5 и 5 ситуация похожа на пороги 5.3 и 10, но образовалось несколько небольших кластеров, "отделившихся" от самого большого в местах выпетливания полипептидной цепи на поверзности белка.
При порогах 4 и 5 уже находятся 18 небольших кластеров, находящихся между элементами вторичной структуры (между альфа-спиралями) и в местах выпетливаний. Вероятно, этот результат можно назвать интерпретируемым, так как эти гидрофобные кластеры находятся практически во всех пространствах между альфа-спиралями.
Для анализа гидрофобных взаимодействий между цепями была взята структура деоксигемоглобина 1FDH . В структуре четыре цепи - А, B, G, H. При запуске ClouD с параметрами 5.3 для порога расстояния и 5 для размера кластера находится 6 кластеров, 2 из которых - большие кластеры самих цепей (не представляют интереса и поэтому не представлены), а оставшиеся 4 располагаются между цепями: 2 между цепями A и H и симметрично 2 между цепями B и G.
d4. Построение поверхности
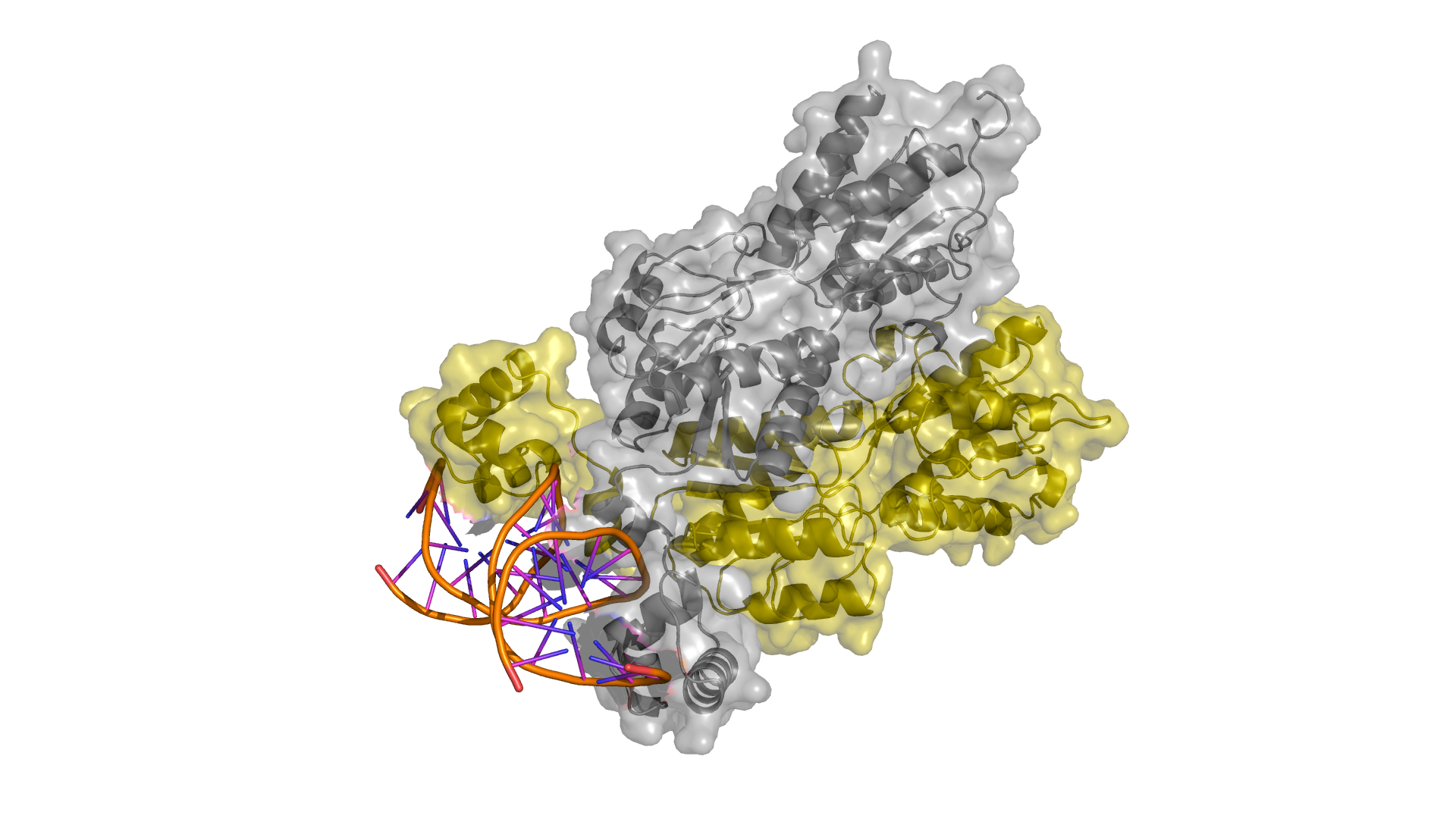
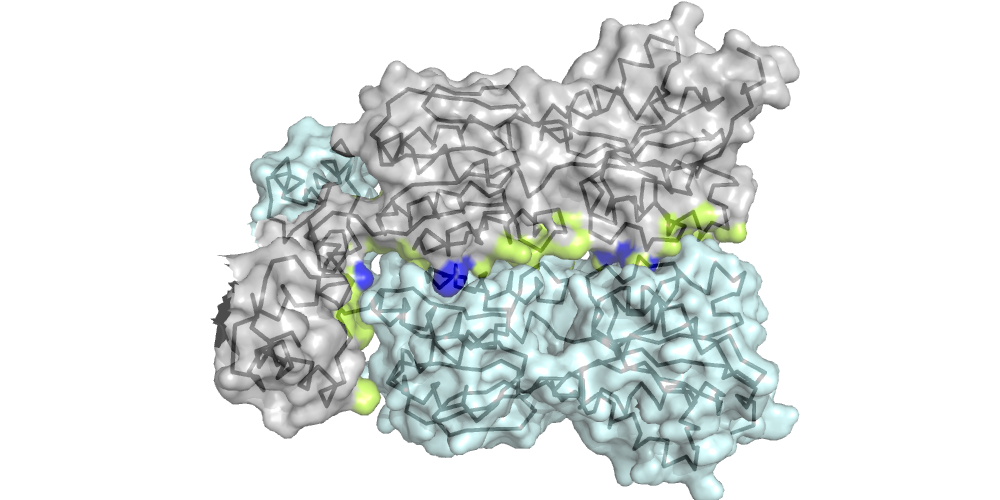
Для комплекса димера пуринового репрессора с ДНК (биологическая единица 2PUD ) с использованием средств программы PyMol были созданы изображения, демонстрирующие контакт мономеров белка между собой и контакт белка с ДНК.
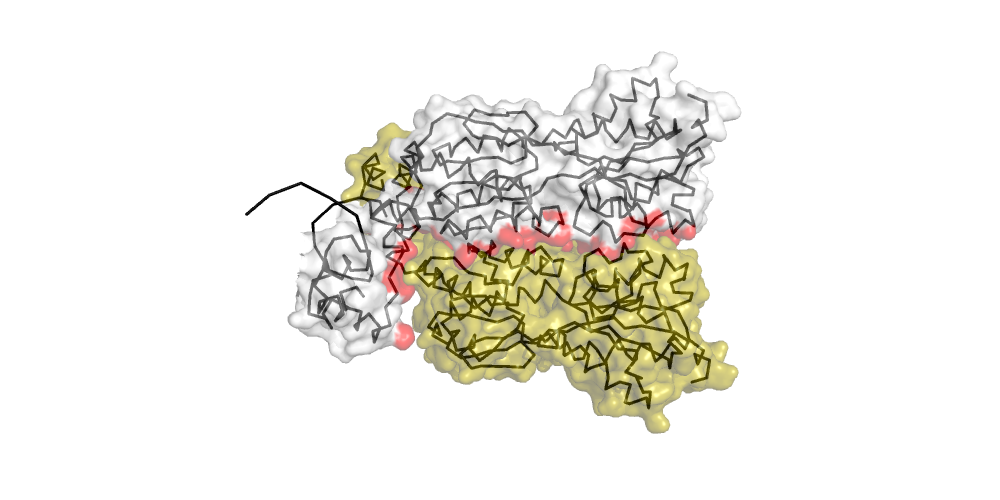
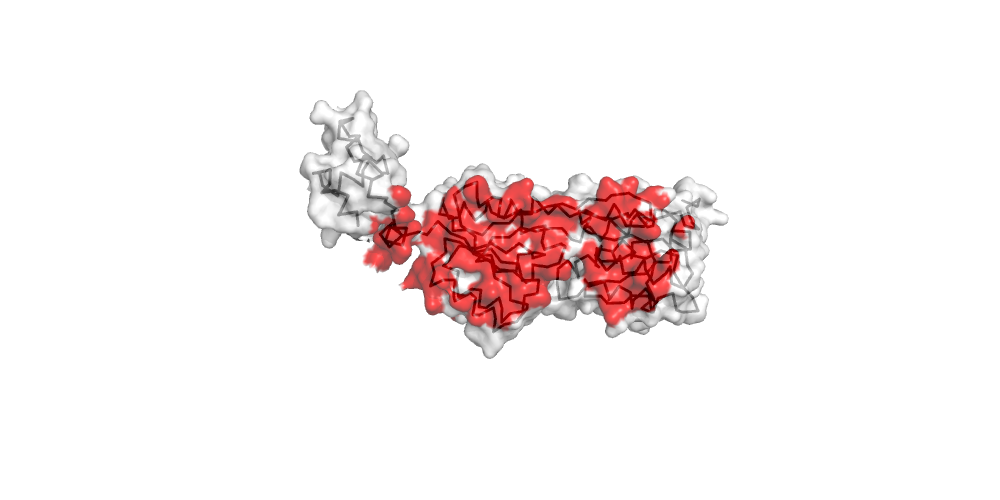
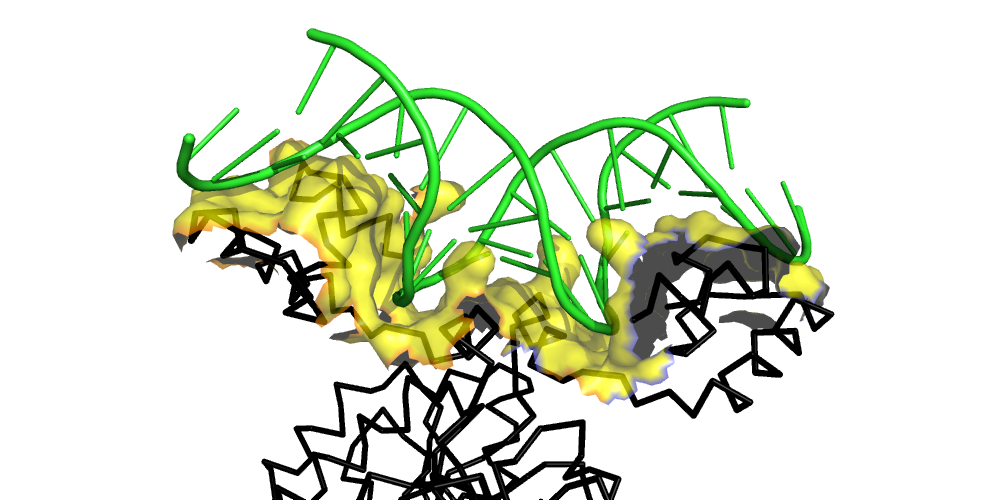

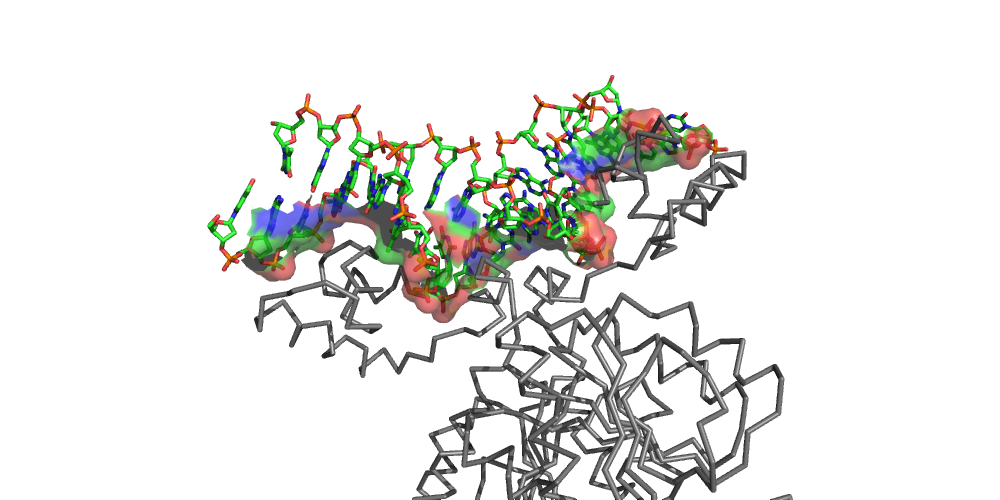
С помощью сервиса Clud в структуре были определены гидрофобные кластеры объемом не менее 10 атомов на интерфейсе мономеров белка (порог на расстояние - 5). Всего нашлось 6 гидрофобных кластеров
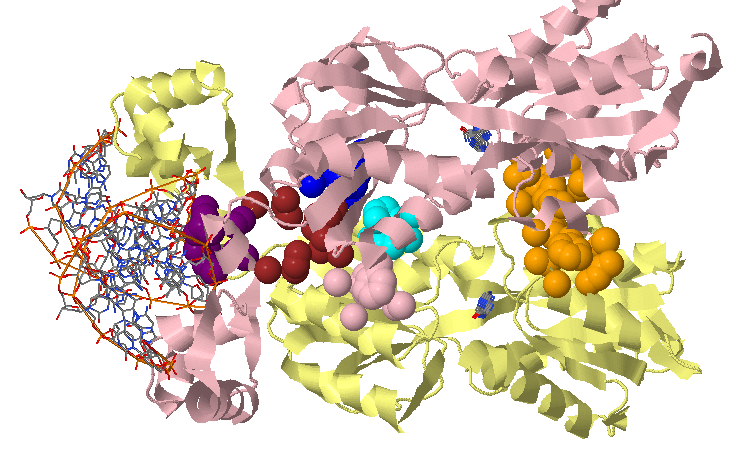
Текстовая выдача Clud . Почему-то в текстовой выдаче содержится также информация о кластерах меньшего размера (меньше 10).
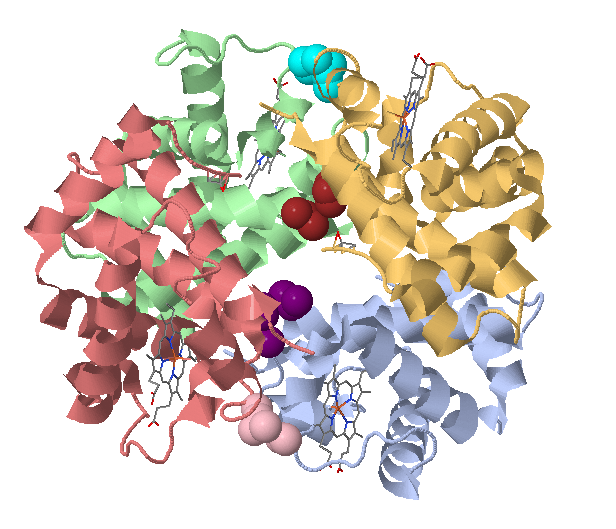
d5. Сравнение доменов SCOP и Pfam
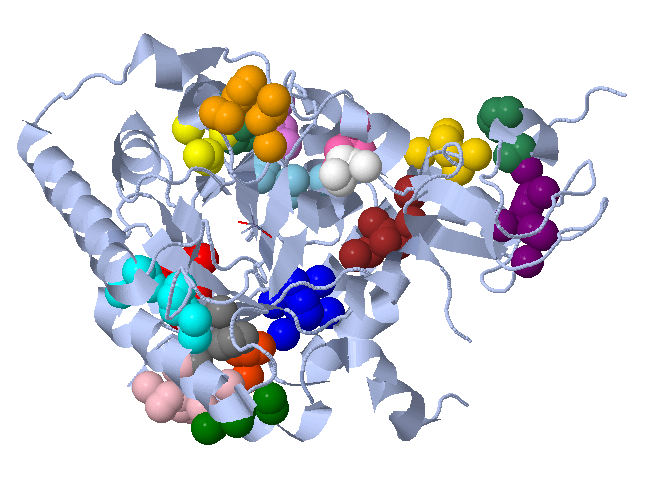
Для структуры 1VLI был произведен поиск доменов в SCOP и Pfam .
Находятся следующие домены:
Pfam: домен NeuB с координатами границ 37 - 276
и домен SAF с координатами границ 304 - 365.
SCOP: домен d1vlia2 с координатами границ 2 - 296
и домен d1vlia1 с координатами границ 297 - 368.
Границы доменов можно обозначить на структуре:
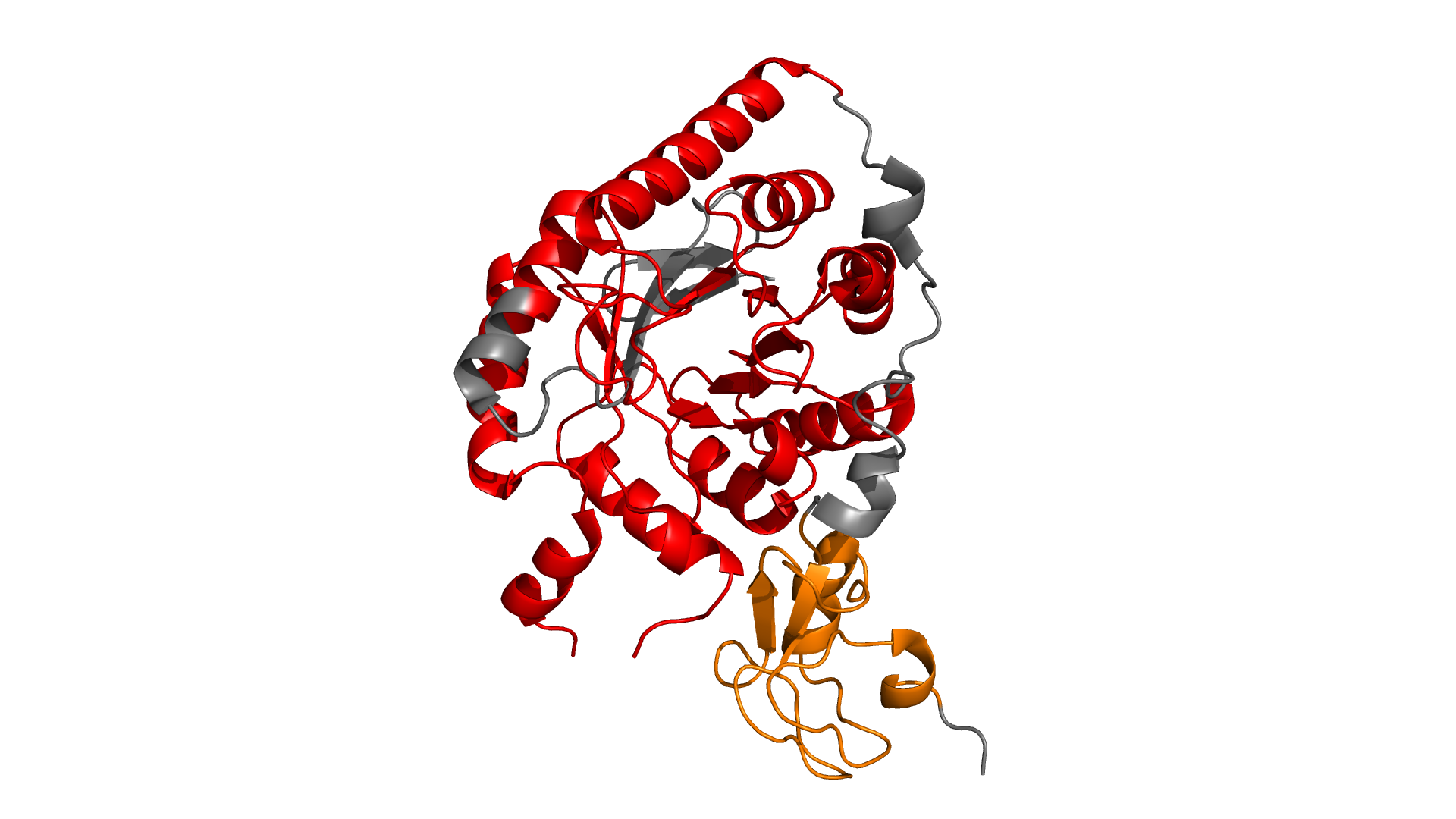
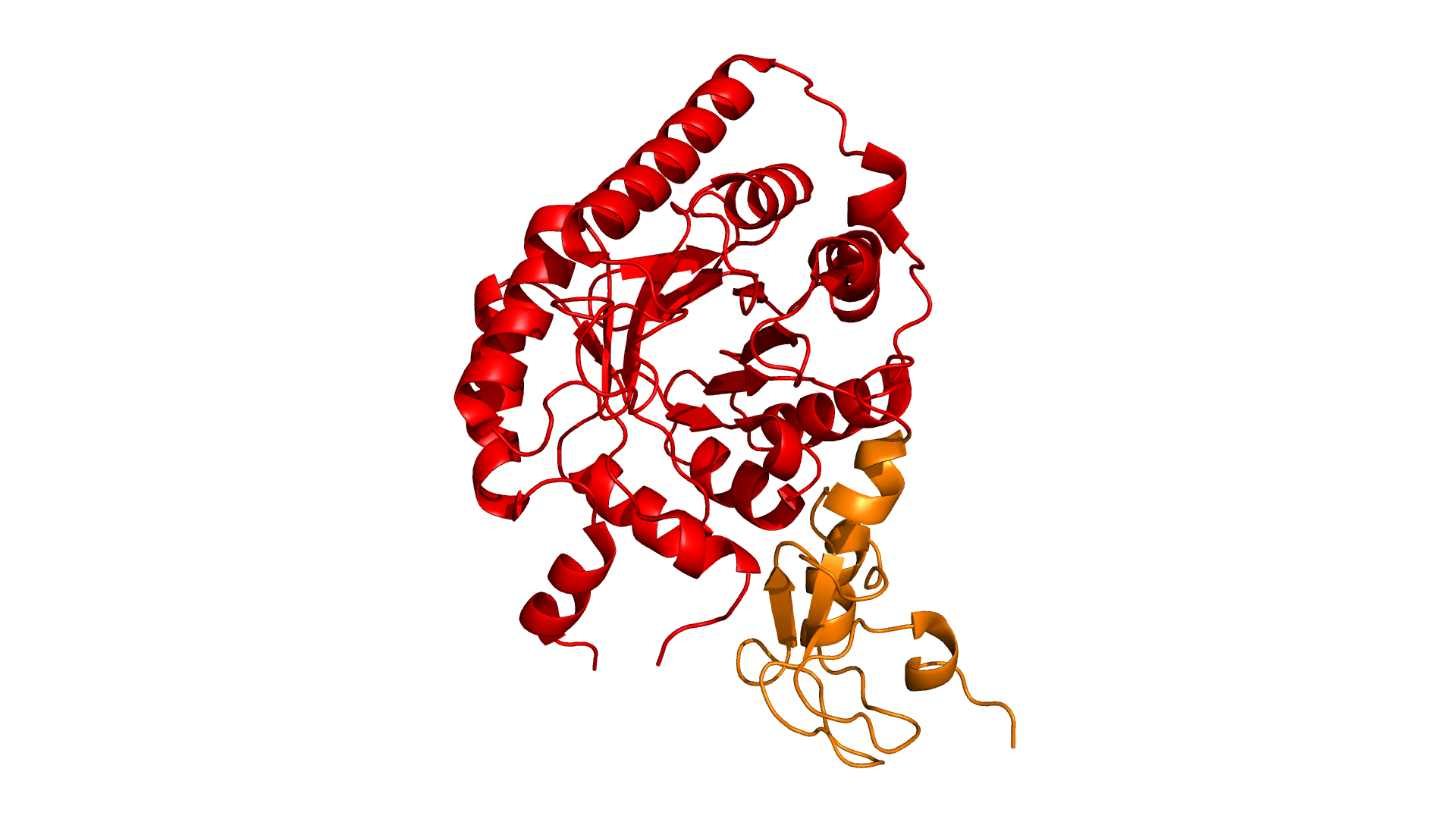
Видно, границы доменов похожи, но у Pfam домены меньше и включены в домены SCOP. Это можно объяснить тем, что в SCOP расположены структурные домены, а в Pfam - функциональные.