1
Был выбран лизоцим из Bacteriophage PS119 (LYS_BPPS1).
C помощью Clustal было построено выравнивание последовательностей выбранного белка и лизоцима форели (1lmp):
1LMP_A|PDBID|CHAIN|SEQUENCE KVYDRCELARALKASGMDGYAGNSLPNWVCLSKWESSYNTQATNRNTDGS 50
sp|O80292|LYS_BPPS1 -----MAMSPALRNSVMAAISGGAIAIASVLITGPGGNDGLEGVRYKPYK 45
:: **: * * . :*.::. * . .. : * . .
1LMP_A|PDBID|CHAIN|SEQUENCE TDYGIFQINSRYWCDDGRTPGAKNVCGIRCSQLLTDDLTVAIRCAK---R 97
sp|O80292|LYS_BPPS1 DVVGVLTVCYGHTGKD-IMPG-KTYTEAECKALLNKDLITVARQINPYIK 93
*:: : : .* ** *. .*. **..** .. * : :
1LMP_A|PDBID|CHAIN|SEQUENCE VVLDPNGIGAWVAWRLHCQNQDLRS-----------------------YV 124
sp|O80292|LYS_BPPS1 VDIPETTRGALYSFVYNVGAGNFRTSTLLRKINQGDIKGACDQLRRWTYA 143
* : . ** :: : ::*: *.
1LMP_A|PDBID|CHAIN|SEQUENCE AG---CGV---------------- 129
sp|O80292|LYS_BPPS1 GGKQWKGLMTRREVERDVCLWGKQ 167
.* *:
2 и 3
Выходной файл в формате .pir был модифицирован для программы modeller: lysozyme.pir
В файле со структурой были удалены все молекулы воды, а всем остаткам лиганда присвоен один номер и соответствующие индексы: 1lmp_now.ent
4 и 5
Был написан скрипт, который говорит modeller построить пять моделейвыбранного белка с лигандом, сохраняя взаимное расположение трех пар атомов белка и лиганда, образующих водородные связи
from modeller.automodel import *
class mymodel(automodel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('N:54:A', 'O7B:168:B'),
('O:103:A', 'N2B:168:B'),
('N:105:A', 'O6C:168:B')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.upper_bound(group=physical.upper_distance,
feature=features.distance(*atoms), mean=3.5, stdev=0.1))
env = environ()
env.io.hetatm = True
a = mymodel(env, alnfile='aligned.pir', knowns=('1lmp'), sequence='seq')
a.starting_model = 1
a.ending_model = 5
a.make()
Запускаем исполнение скрипта командой
mod9v7 lys_bpps1.py &После запуска скрипта мы получили пять моделей белка с лигандом.
6
Рассмотрим полученные модели
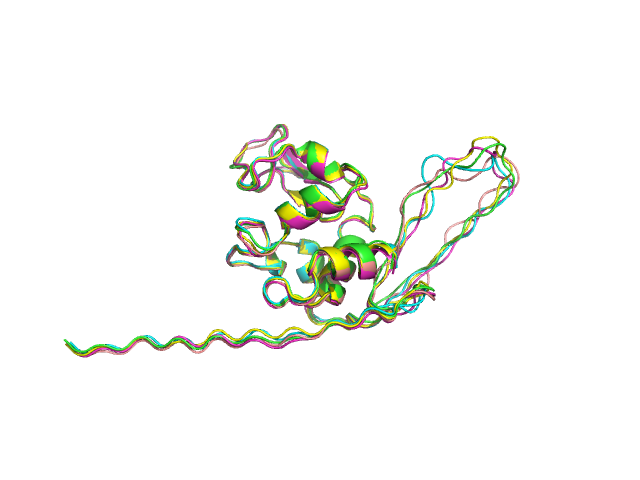
Модели очень похожи. Расхождения в петлях и N-конце (не структурирован и представлен прямой цепью, так как в референсной структуре его не было)
7
Качество моделей проверялось WHATIF.
| Model 1 | Model 2 | Model 3 | Model 4 | Model 5 | |
| Ramachandran Z-score | -0.810 | -1.137 | -1.264 | -1.288 | -1.267 |
| RMS Z-score for bond lengths | 0.988 | 0.975 | 0.985 | 0.985 | 0.999 |
| RMS Z-score for Improper dihedralbond angles | 1.312 | 1.295 | 1.272 | 1.338 | 1.319 |
| RMS Z-score for Improper dihedral | 1.104 | 1.121 | 1.095 | 1.052 | 1.046 |
Лучшая модель по z-score Рамачандрана - 1; по Bond lengths - 5; по Bond angles - 3; по Improper dihedral - 5. Если оценивать по этим критериям, то 5 модель кажется лучше остальных.