Вторичная структура tRNA. DNA-белковые комплексы
Предсказание вторичной структуры заданной tRNA
В этом задании нужно было сравнить выдачи программ einverted, RNAfold и ранее
использованной find_pair для предсказания вторичной структуры RNA.
Последовательность RNA для einverted была получена со страницы PDB и записана в файл 1f7u_trna.fasta.
Для корректной обработки нуклеотид I был заменён на X, а название последовательности было удалено.
Далее была исполнена следующая команда:
einverted 1f7u_trna.fasta -gap 0 -thr 1 -match 1 -mis -1 -outfile emboss_001.inv -outseq emboss_001.fasta
Затем была использована команда RNAfold,а также
веб-сервис для визуализации:
export PATH=${PATH}:/home/preps/golovin/progs/bin
Полученные файлы можно посмотреть здесь:
cat 1f7u_trna.fasta | RNAfold --MEA -p > zuker.rna
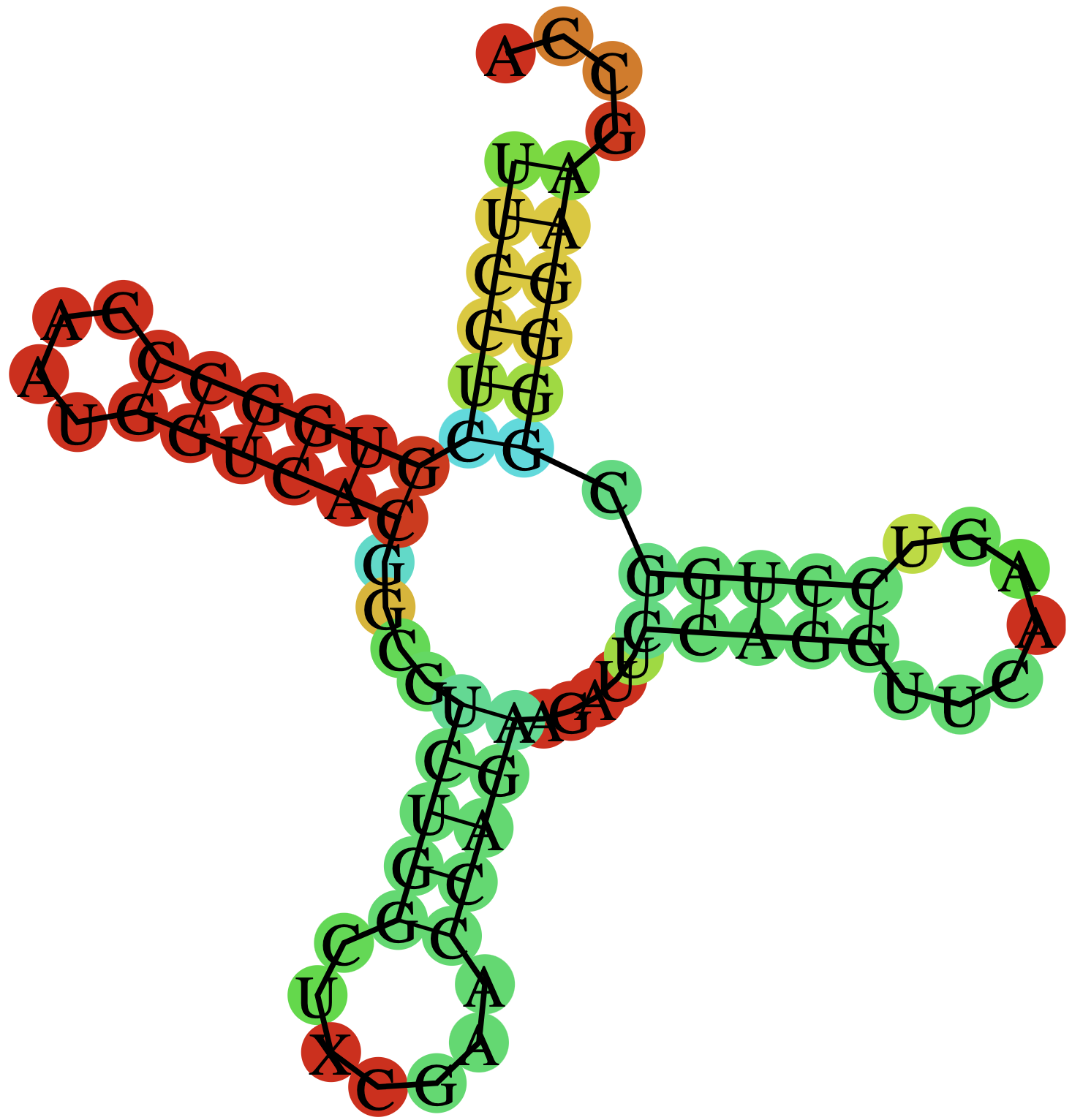
Таблица 1. Параметры разных структур DNA
| Участок структуры | Позиции в структуре (find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера (RNAfold) |
| Акцепторный стебель | 5'-901-907-3' 5'-966-972-3'Всего 7 пар, из них 5 канонических |
5 | 5 пар и 1 лишняя |
| D-стебель |
5'-910-913-3' 5'-922-925-3'4 пары, из них 3 канонические |
- | 3 пары и 3 лишних |
| T-стебель |
5'-949-953-3' 5'-961-965-3'5 канонических пар |
- | 5 |
| Антикодоновый стебель |
5'-939-944-3' 5'-926-931-3'6 пар, из них 4 канонические |
4 и 1 лишняя | 4 и 1 лишняя |
| Общее число канонических пар нуклеотидов | 19 | 9 | 17 |
Поиск DNA-белковых контактов в заданной структуре
Для работы во втором задании мне был выдан pdb файл 1TRO. Но nucplot (см. далее) не работал, поэтому я выбрал другой белок - 1HW2. В начале нужно было вспомнить, как с помощью команды define JMol задавать множества атомов, затем:
- определить множество атомов кислорода 2'-дезоксирибозы (set1);
- определить множество атомов кислорода в остатке фосфорной кислоты (set2);
- определить множество атомов азота в азотистых основаниях (set3);
- создать скрипт-файл с определениями этих множеств;
select, чтобы в выводе увидеть количество нужных атомов.
Посмотреть файлы можно здесь:
Результат представлен в таблице 2.
Таблица 2. DNA-белковые контакты
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 3 | 22 | 25 |
| остатками фосфорной кислоты | 6 | 5 | 11 |
| остатками азотистых оснований со стороны большой бороздки | 1 | 1 | 2 |
| остатками азотистых оснований со стороны малой бороздки | 5 | 10 | 15 |
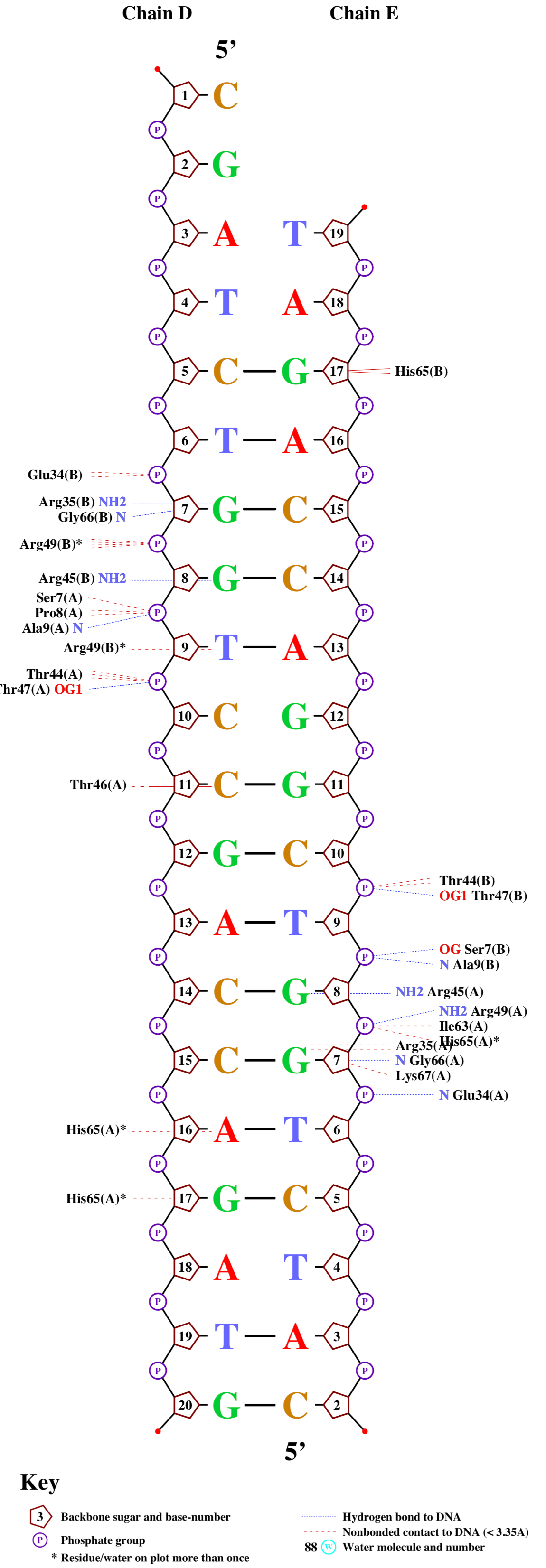
Далее нужно было получить схему ДНК-белковых контактов.
Для этого я перевёл его в старый формат записи и воспользовался nucplot:
remediator --old ''1hw2.pdb'' > ''1hw2_old.pdb''
Полученное изображение я перевел в pdf и совместил в фото-редакторе. Результаты на рис. 2.
nucplot 1hw2_old.pdb
На полученной схеме нужно было выбрать и показать с помощью Jmol:
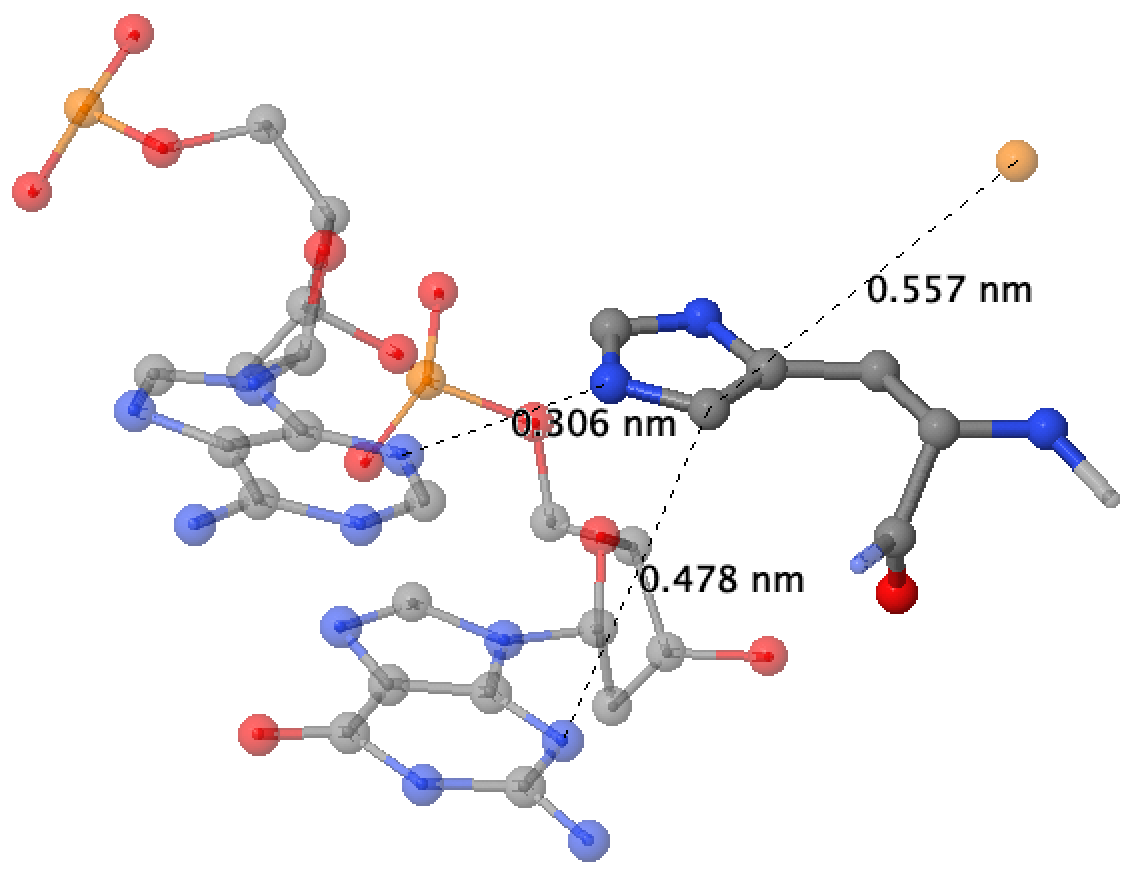
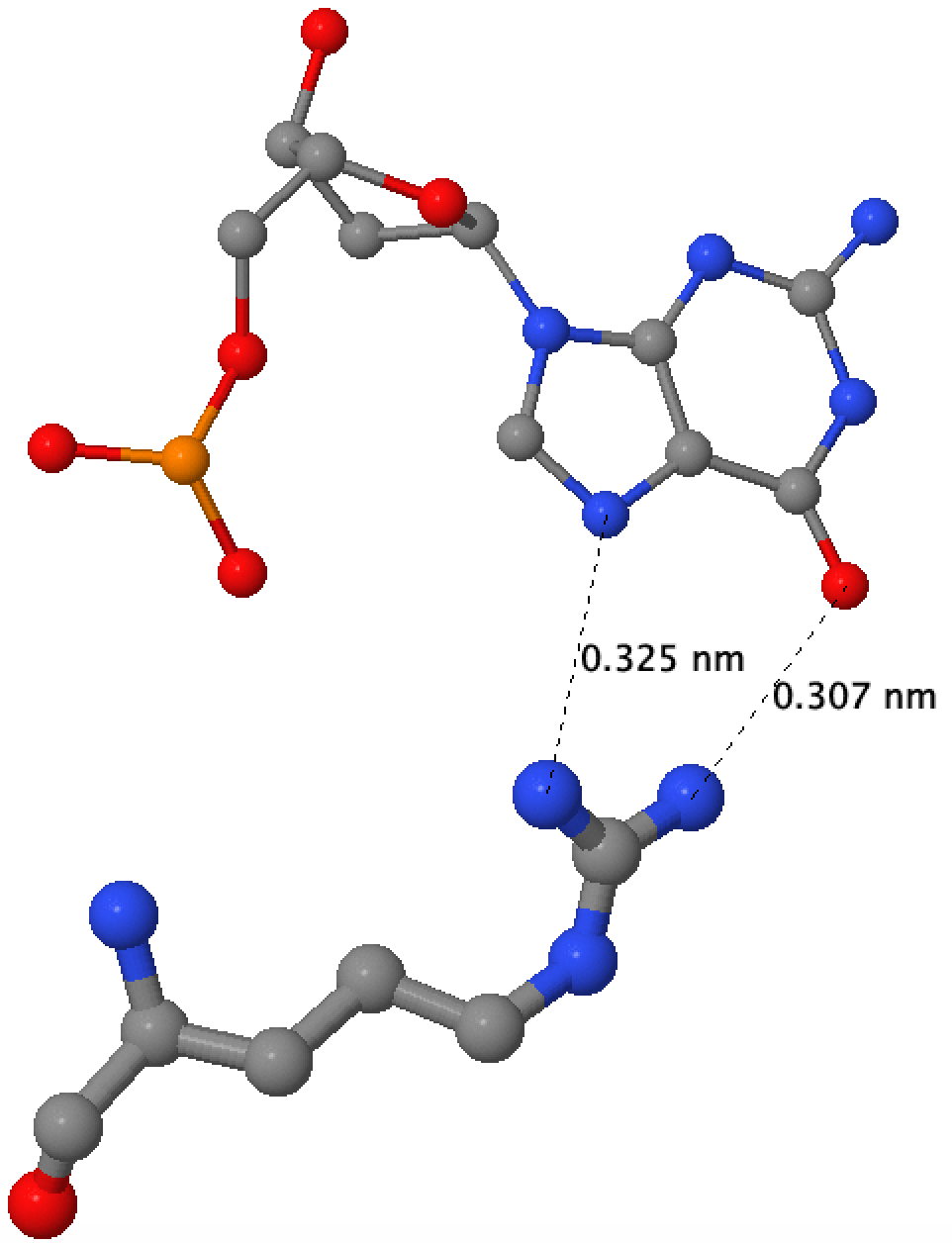
- Аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК - это His65 цепи A. На рисунке его легко найти, потому что он помечен звездочкой. Видно, что он образует связи с A16, G17 и фосфатной группой G8.
- Аминокислотный остаток, по моему мнению, наиболее важный для распознавания последовательности ДНК. Я считаю, что это Arg35 цепи A, поскольку эта аминокислота в двух местах связывается с нуклеотидом, а судя по pdb-модели, делает это в районе большой бороздки, по которой и проходит молекулярное узнавание.
Сами контакты были визуализированны с помощью JMol.