
эволюции гена

| GO TO: |
|
Описание исходного дерева |
||||||||||||||||||||||||||||||||||||||||
|
Выданное мне дерево в виде скобочной структуры записывается следующим образом: (((А:60,B:80):20,(C:50,D:50):10):15,(E:80,F:80):5).
На основе этого мною был реконструирован его внешний вид: 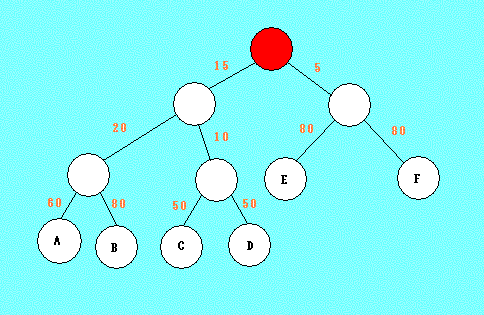
В виде разбиения множества листьев это дерево выглядит следующим образом:
|
||||||||||||||||||||||||||||||||||||||||
|
Моделирование мутантных последовательностей, соответсвующих дереву Построение правдоподобных деревьев |
||||||||||||||||||||||||||||||||||||||||
Для построения мутантных последовательностей, заданного мне гена белка HEMN_ECOLI, соответвующих данному дереву
исползьзовался скрипт следующего вида:msbar hemn.fasta abcd.fasta -point 4 -count 274 -auto msbar abcd.fasta ab.fasta -point 4 -count 365 -auto msbar ab.fasta a.fasta -point 4 -count 1096 -auto msbar ab.fasta b.fasta -point 4 -count 1461 -auto msbar abcd.fasta cd.fasta -point 4 -count 183 -auto msbar cd.fasta c.fasta -point 4 -count 914 -auto msbar cd.fasta d.fasta -point 4 -count 914 -auto msbar hemn.fasta ef.fasta -point 4 -count 91 -auto msbar ef.fasta e.fasta -point 4 -count 1462 -auto msbar ef.fasta f.fasta -point 4 -count 1462 -autoСкрипт представляет собой последовательность запусков программы msbar пакета EMBOSS, на каждом этапе создаётся одна из последоветльностей стоящих в узлах дерева. Значения -count посчитаны по формуле N=1.33*S*a/100 где S - длина моего гена - 1374нт a - расстояние по соответствующей ветви дерева - нормированное на сто количество мутаций между последовательностями. Коэффициент 1.33 = 4/3 - поправка на то, что реальное количество замен, произошедших в последовательности в среднем 4/3 раза больше чем количество замен, приведших к изменению последовательности. Далее на основе полученных последовательностей-листьев были построены новые деревья алгоритмами максимального правдоподобия, Neighbor-joining и UPGMA: Предварительно был создан файл leafs.fasta с "выравниваниями". Затем программой fdnaml было создано дерево алгоритмом максимального правдоподобия: fdnaml leafs1.fasta -ttaratio 1 -auto, где -ttratio - это отношение транзиций к трансверсиям. Для выполнения следующих реконструкций дерева, были подстчитаны попарные расстояния между последовательностями: fdnadist leafs1.fasta -ttratio 1 -auto Используя полученный файл, была проведена реконструкция дерева алгоритмом Neighbor-joining: fneighbor leafs1.fdnadist -auto, (выходной файл был переименован в NJ_t.fneighbor) и алгоритмом UPGMA fneighbor leafs1.fdnadist -outfile UP_t.fneighbor -treetype u -auto I. Nucleic acid sequence Maximum Likelihood method, version 3.6b +----------------------------B | | +------------------------F | +--------4 | | +------------------------E 1-------3 | | +-----------------D | +----2 | +---------------C | +---------------A Выходной файл: M_L_M II. Neighbor-Joining/UPGMA method version 3.6b Neighbor-Joining method +------------------------------B ! ! +--------------C ! +-----3 ! ! +------------------D 1----------4 ! ! +------------------------E ! +---------2 ! +-------------------------F ! +--------------A Выходной файл: N_J_M III. Neighbor-Joining/UPGMA method version 3.6b UPGMA method +----------------------A +----2 ! +----------------------B +---4 ! ! +----------------C ! +----------1 --5 +----------------D ! ! +-------------------------E +-----3 +-------------------------F Выходной файл: UP_M Для сравнения составим сводную таблицу ветвей:
Как видно из этой таблицы все три алгоритма воссоздали исходные деревья. Стоит так же отметить, что UPGMA создал укоренённое дерево. |
||||||||||||||||||||||||||||||||||||||||