Структура РНК
| GO TO: |
| Общие данные. | ||||
| Выданная мне структура 1h4q (PDB code) включает в себя белок класса E.C.6.1.1.15 - пролил-тРНК синтазу (Prolyl-tRNA synthetase) - (chains A and B)и пролиновую тРНК(cgg) (Trnapro(cgg)) (chain T). Обе макромолекулы относятся к Thermus Thermophilus штамма HB8. В структуре так же присутсвуют молекула ATP и молекула пролинола. | ||||
| Данные о структуре моей tRNA | ||||
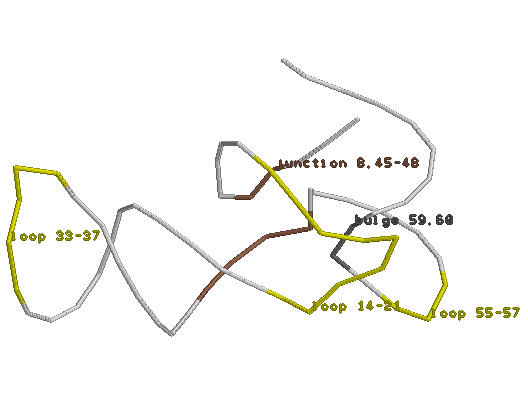
Ниже приведены изображение структуры и последовательность моей пролиновой тРНК с указанием спиралей и номеров остатков, а так же участков между ними:
Для сравнения: если искать возможные двуспиральные участки в подобной структуре, анализируя её нуклеотидную последовательность, опираясь только на Уотсон-Криковскую теорию спаривания комплементарных оснований - то есть, просто ища взаимно комплементарные (или почти комплементарные) инвертированные кусочки в этой структуре, то получается следующий результат:
FIND_PAIR же работает исключительно с PDB-структурами и предсказывает спирали, основываясь на расстояниях между основаниями, не обращая внимания на их (не)комплементарность, что судя по картинке даёт более жизненный результат. (NOTE: из 26 пар, выделенных FIND_PAIR в спирали, только 7 были неканоническими). Так же стоит отметить два модифицированных основания в моей тРНК: 54-ое - обозначенное как 5MU - видимо "М"утировавший в "5"-ом положении "У"ридин - по существу же просто Тимин; и 55-ое - псевдоуридин. Здесь лежит скрипт, по которому строились продемонстрированные выше картинки, за исключением вращения и загрузки файла. В моей тРНК мной были так же найденны внеспиральное стекинг-взаимодействие между основаниями, и внутриспиральные не Уотсон-Криковские взаимодейтвия, как изображено ниже 
Стекинговое взаимодействие 
Не Уотсон-Криковские взаимодействия. 
|
||||
| Сравнение структур тРНК и спирализованной ДНК | ||||
|
Сравнивать каждую их 4-ех спиралей с той или иной формой ДНК мне показалось несостоятельным, ибо,
с одной стороны, "по виду" они все напоминают А-форму: - они "толстые", большая бороздка уже маленькой, и т.д.,
а с другой стороны, каждый стебель слишком мал, что бы делать обоснованные выводы.
А поэтому для более определенного доказательства я объединил красную и оранжевую спирали в
"первую спираль", а синюю и зелёную спирали - во "вторую спираль". Это логично - из общего вида
моей тРНК, и согласуется с данными, выдаваемыми программами FIND_PAIR и ANALYZE, которые выделяют
как раз именно эти спирали - как две основные (эти программы, как уже было сказано выше, работают
в основном с расстояниями в трёхмерной структуре, а потому понимают под спиралями нечто иное, нежели
просто "стебли").
Для сравнения так определённых спиралей мною была измерена ширина бороздок, а так же померены торсионные углы остатков 1-ой и 2-ой спиралей с помошью программы ANALYZE:
1) Для превой спирали: Минимальная ширина большой бороздки - померена от нуклеотида C61 - составила 12.42 ангстрема, максимальная - от того же нуклеотида - 14.605 ангстрема. Средняя ширина малой бороздки составила 15.201 ангстрема. - Видно что малая бороздка шире, что свойственно A-форме. 2) Для второй спирали: Ширина второй бороздки составила 8.68 ангстрема Ширина малой бороздки составила 16.85 ангсрема. - Соотношение ширин бороздок так же больше свойственное A-форме. 3) Диаграмма торсионных углов: 
Данная диаграмма демонстрирует, что на самом деле ни одна из спиралей не представляет из себя чистой A- или Б- формы. Однако для воторой спирали хорошо видно, что по углам α, γ, δ, ζ, χ, - основным определяющим углам этих форм (как уж отмечалось в предыдущем практикуме, угол β в "живых" структурах, похоже, очень варьируется, а угол ε в обоих формах очень близок) - она заметно ближе к A-форме. Тоже можно сказать и про первую спираль, но только в случае углов α, δ и ζ - что затрудняет её определение как той или иной формы НК. Опеределение конкретной формы для всей тРНК невозможно, ибо она имеет нерегулярную, изломанную структуру, и включает, как было показано выше, несколько стеблей, в той или иной похожих друг на друга, формирующих и более сложные спирали. Подобная нерегулярность структуры так же может объясняться и тем, что мы имеем дело со структурой не чистой тРНК, а находящейся во взаимодействии с белком - пролин-тРНК синтазой, основной функцией которой, судя по описанию, является присоединения к тРНК аминокислоты. В ходе это реакции тРНК возможно деформируется под воздействием связывающих доменов белка. |
||||
| Предсказание вторичной структуры тРНК с помошью программы MFOLD | ||||
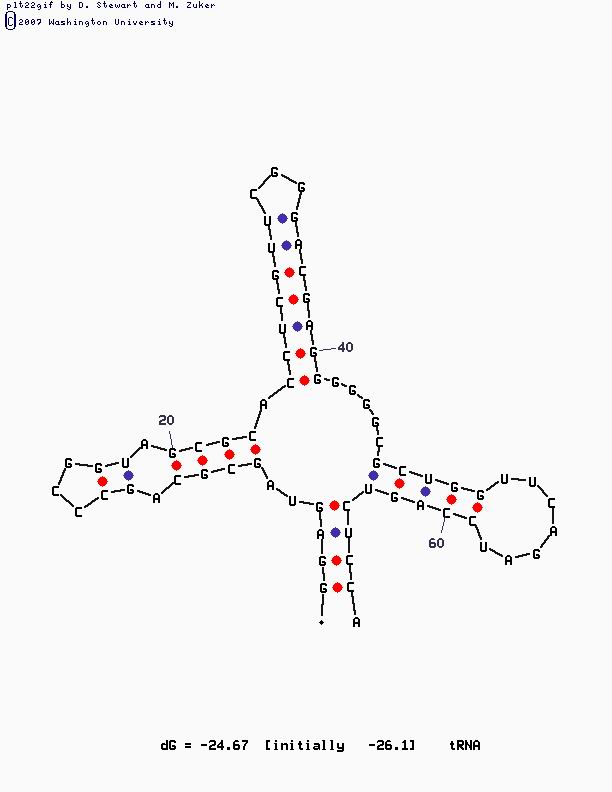
С помощью программы MFOLD мной было полученно несколько структур. Предсказание проводилось c параметрами P=15, P=20, P=30. Наилучшей, но далеко не идеальной оказалась вторая структура при P=15:

Достоинством это структуры является почти совпадающие с найденными мною красным и оранжевым стеблями нижний и правый стебли этой конструкции, наличие мультипетли в центре структуры и более или менее похожие на обозначенные ранее синие и зелёные стебли - соотвественно верхний и левый стебли предсказанной mfold-ом структуры. Получить лучшего предсказание не удалось. | ||||