Раздел EMBOSS
Можно обращаться к отдельной странице, посвященной не только командам пакета EMBOSS, но и к описанию некоторых особенностей командной оболочки Bash.
– команда биоинформатического пакета EMBOSS (European Molecular Biology Open Software Suite).
Команда
выводит в консольное окно информацию о возможностях infoseq. Результат указанной команды – англоязычное описание различных параметров (или квалификаторов) для infoseq.
В файле infoseq_help.txt находится это описание. Отметим, что, т. к. информация о командах с –help выводится в стандартном потоке ошибок, получить ее можно с помощью команды
которая выводит информацию и stdout, и stderr.
Различные параметры команды infoseq будут рассмотрены далее.
I. Стандартные (или обязательные) параметры.
В данном случае это параметр “-sequence”. Вывод информации с этим параметром аналогичен выводу информации без него. То есть равносильны команды
и их результат есть информация SwissPtrot о белке PDXS_BACSU в файле seq.txt.
II. Продвинутые параметры.
1. “-columns” – параметр, представляющий информацию в виде таблицы с колонками, как в файле seq.txt.
2. “-no” – параметр пишется слитно c другим. На экран выводится отрицание последнего. Так команда
дает результат из файла nocolumns_seq.txt.
3. “delimiter” – параметр, с помощью которого данные разделяются заданным символом (символ задается после параметра в двойных кавычках). Например, результатом команды
будет файл delimiter_seq.txt
4. “(no)heding” – параметр с (no), с использованием которого информация о последовательности выводится в виде таблицы без заголовков. Например, с помощью команды
можно получить результат из файла noheading_seq.txt.
5. “-only” – параметр, с помощью которого можно выбрать для вывода на экран определенные данные о последовательностях белка.
Используется со следующими параметрами:
“-database” – название базы данных, из которой берется информация;
“-name” – имя объекта (белка);
“-accession” или “AC” – код доступа;
“-gi” – особый номер, который присваивается последовательностям из баз данных NCBI (National Center for Biotechnology Information);
“-type” – тип белка;
“-length” – количество аминокислотных остатков;
“-pgc” (percent G-C content) – процент G-C пар оснований;
“-organism” – организм, которому принадлежит белок;
“-description” – описание.
Например, если необходимо получить информацию только о названии белка и его коде доступа (accession number), то можно воспользоваться командой
результат в файле only_seq.txt
III. Опциональные (или дополнительные) параметры.
1. “-outfile” – записывает информацию о последовательностях в заданный файл.
2. “-html” - выводит html-код, при записи которого на web-сайт получается следующая таблица:
| USA | Database | Name | Accession | Type | Length | Organism | Description |
|---|---|---|---|---|---|---|---|
| sw-id:PDXS_BACSU | sw | PDXS_BACSU | P37527 | P | 294 | Bacillus subtilis (strain 168) | Pyridoxal biosynthesis lyase PdxS (4.-.-.-) (Superoxide-inducible protein 7) (SOI7) |
IV. Общие параметры.
1. “-help” – выводит на экран некоторые параметры для командной строки;
2. “-verbose” – выводит на экран подробную информацию о параметрах для командной строки (больше, чем “-help”).
Использование этих команд приведено выше.
С помощью команды infoseq можно получать информацию сразу о многих последовательностях.
Пример. При желании узнать описание (в которое включены функции) белка PDXS у всех известных видов бактерий рода Bacillus, можно воспользоваться командой
результат – таблица в файле decr_pdxs.txt. Правда, описание белка везде одинаковое, что не удивительно.
То же можно использовать при поиске информации о всех белках, имеющихся в базе данных, одной бактерии и т.п.
– команда биоинформатического пакета EMBOSS.
Аналогичный для infoseq синтаксис имеет команда entret. Entret прежде всего используется для быстрого получения информации о последовательностях из различных баз данных. Так, например, с помощью команды
можно получить информацию о белке PDXS_BACSU из базы данных Swiss-Prot. Команда по умолчанию запрашивает у пользователя полное имя файла, в который необходимо поместить информацию. В консольном окне это будет выглядеть следующим образом:
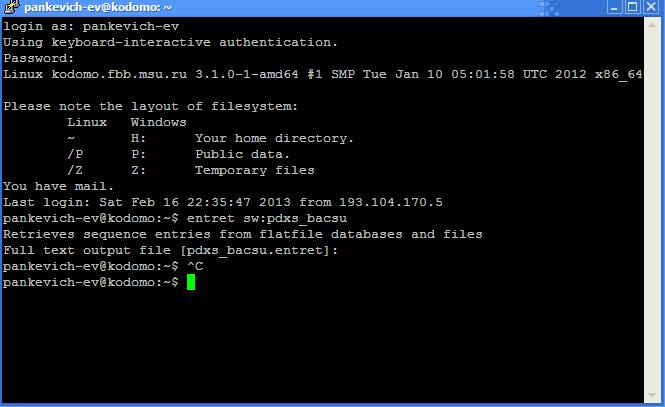
В квадратных скобках при этом указывается имя в файла, в которой будет загружена информация, если пользователь не укажет другое название. Так в данном случае описание белка PDXS_BACSU можно увидеть в файле pdxs_bacsu.entret