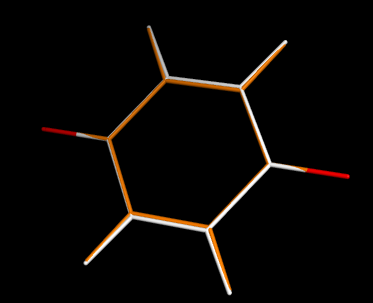
Из файла 1.smi со SMILES порфирина программой из babel был создан файл 1.mol с 3D структурой порфирина, в PyMol структура выглядит так (1.pdb после удаления двух лишних водородов):
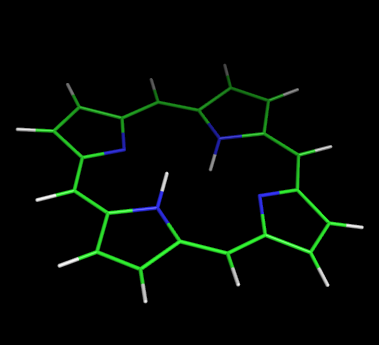
С помощью babel координаты в mol формате переформатированы во входные файлы для Mopac:- с параметризацией типа PM6 1_opt.mop
- с параметризацией типа AM1 1_opt_AM1.mop
Выходные файлы Mopac и созданные из них pdb:- PM6 1_opt.out и 1_opt.pdb
- AM1 1_opt_AM1.out и 1_opt_AM1.pdb
Стуктуры, полученные с разной параметризацией, на картинке ничем не отличаются: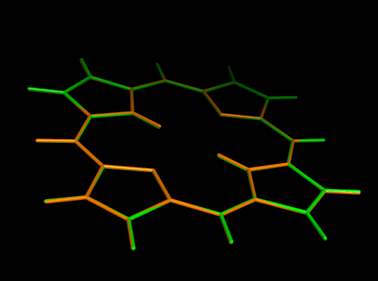
В файлах *.out различия есть - различные использованные матрицы Гессе, рассчитанные теплоты образования, энергии, заряды атомов и пр. Рассчитанные координаты так же различны, но, видимо, не настолько значительно, чтобы это было заметно при графическом изображении.
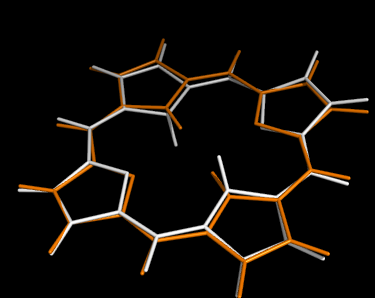
Структуры, полученные через программы Mopac и пакета babel и, видимо различаются: первая плоская (оранжевый), в то время как вторая - не совсем (белый):

Файл с рассчитанными возбужденными состояниями порфирина: 1_opt_spectr.out. Длина волн, при которых происходят переходы на уровни:
Уровень Энергия, эВ Длина волны, нм 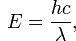
1 0 0 2 1,913686 647,88 3 2,266217 547,10 4 2,462868 503,41 5 2,824367 438,98 6 3,362519 368,72 7 3,390031 365,73 8 3,669386 337,89 9 3,871186 320,27 Для молекулы O=C1C=CC(=O)C=C1 определена геометрия как с помощью obgen 2.pdb, так и Мopac 2_opt.out 2_opt.pdb.
Для той же молекулы с зарядом -2: 2_opt_ch.out 2_opt_ch.pdb.
Структуры, полученные с помощью babel (голубой) и Mopac (белый) не различимы:

Результаты сравнения файлов *.out- Энергии образования, суммарные энергии, электронные энергии различаются, но не сильно
- Значительно различаются ионизационные потенциалы: 10.593801 EV для незаряженной и -2.140430 EV для заряженной молекул.
- Обе структуры содержат 12 атомов, включая 4 атома водорода - т. е. в заряженной молекуле протоны все равно присутствуют
- Координаты различаются в основномтолько во втором знаке
- Как и следовало ожидать, заряды, особенно на кислороде, различны
- Различное распределение электронной плотности (atomic orbital electron populations)
- суммарное значение диполей одинаковое, но различно распределено по осям, что отражается на структуре - заряженная молекула чуть "сплющена" к оси, соединяющей кислороды.
В PyMol видно, что структуры плоские, различия между незаряженной (белый) и заряженной (оранжевый) есть - в длине некоторых связей (заряженная молекула сжата к оси кислородов из-за другого распределения диполя) - но не очень большое: