SMILES для NAG: nag.smi.
C помощью obgen получаем mol-файл nag.mol, затем конвертируем в pdb-файл nag1.pdb.
Скриптами prepare_ligand4.py и prepare_receptor4.py из пакета Autodock tools создаём pdbqt файлы лиганда и белка, соответственно.
prepare_ligand4.py -l nag1.pdb prepare_receptor4.py -r 2ihl_5.pdb
Полученные файлы: nag1.pdbqt, 2ihl_5.pdbqtТеперь надо создать файл с параметрами докинга vina.cfg.
Для докинга необходимо указать область структуры белка в которой будет происходить поиск места для связывания. Удобно его задать как куб с неким центором. Координаты центра мы определим из модели комплекса, которую мы построили на прошлом занятии. Координаты атома сахара, который по моему мнению находится в центре сайта связывания:HETATM 1029 N2B NAG B 130 40.856 42.108 26.501
Построим файл vina.cfg с таким содержанием:center_x=40.856 center_y=42.108 center_z=26.501 size_x = 25 size_y = 25 size_z = 25 num_modes = 20
Теперь можно провести первый докинг:
vina --config vina.cfg --receptor 2ihl_5.pdbqt --ligand nag1.pdbqt --out nag_prot.pdbqt --log nag_prot.log
Просмотрим файл nag_prot.log. Энергии 3ёх лучших расположений и геометрическая разница между ними:mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.6 0.000 0.000 2 -5.5 3.351 5.389 3 -5.5 1.829 4.915В PyMol загрузим файлы nag_prot.pdbqt и 2ihl_5.pdbqt, отобразим все состояния на одной картинке: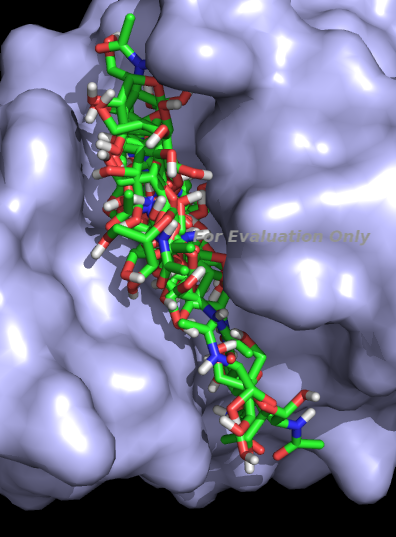
Видно, что лиганд может располагаться вдоль всей "бороздки" в белке.Теперь проведём докинг, рассматривая подвижность некоторых боковых радикалов белка. Сначала разобьем белок на две части, подвижную и неподвижную. Для подвижной части выберем 3 аминокислоты которые использовали в прошлом задании для позиционирования лиганда.
prepare_flexreceptor4.py -r 2ihl_5.pdbqt -s ASN46_ALA107_ARG112
и проведём докинг:vina --config vina.cfg --receptor 2ihl_5_rigid.pdbqt --flex 2ihl_5_flex.pdbqt --ligand nag1.pdbqt --out vina_2ihl_flex.pdbqt --log vina_2ihl_flex.log
Время счета больше, чем в обычном докинге.Энергии 3ёх лучших расположений и геометрическая разница между ними. Афинность снизилась по сравнению с обычным докингом.
mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.2 0.000 0.000 2 -5.1 1.590 1.904 3 -5.0 1.639 3.101В PyMol загрузим файлы nag_2ihl_flex.pdbqt и 2ihl_5_rigid.pdbqt, отобразим все состояния на одной картинке: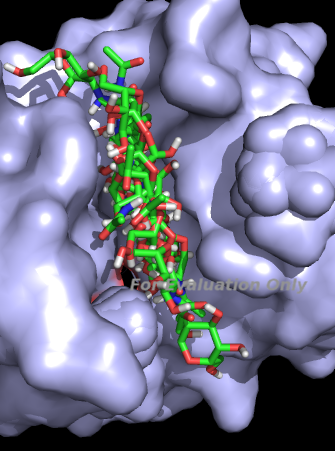
Здесь, по сравнению с обычным докингом, лиганд может занимать больше положений.В результатах докинга лиганд иногда располагается примерно в том же месте, чт оодин из мономеров в результатах моделирования (если смотреть по кольцу), но всесгда в другой ориентации, т. е., скорее всего, связывается за счет других связей. Скорее всего, неточность связана с тем, чт ов моделировании использовался тример лиганда, а в докинге - мономер.
NAG содержит в себе СH3C(=O)NH группу. Созданы 3 лиганда где метильный радикал этой группы будет заменён на OH (nag_o.smi), NH2 (nag_n.smi), H (nag_.smi). Для каждого из этих лигандов проведен обыкновенный докинг, результаты в виде таблицы из трёх лучших расположений для каждого лиганда:
-OH mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.9 0.000 0.000 2 -5.8 2.208 3.709 3 -5.6 2.080 5.087-NH2 mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.8 0.000 0.000 2 -5.7 2.499 4.382 3 -5.5 2.581 5.141-H mode | affinity | dist from best mode | (kcal/mol) | rmsd l.b.| rmsd u.b. -----+------------+----------+---------- 1 -5.2 0.000 0.000 2 -5.0 1.973 3.289 3 -4.9 2.259 3.856Лучшая афнность - у лиганда с -OH вместо метильной группы, худшая - у лиганда с -H. У всех кроме -H афинность лучше, чем у исходного лиганда.