Построим выравнивание последовательности из структуры ID: 1lmp и белка 2IHL (LYSC_COTJA). Будем использовать Clustal и GeneDoc.
Полученное выравнивание в формате PIR: LYSC.pir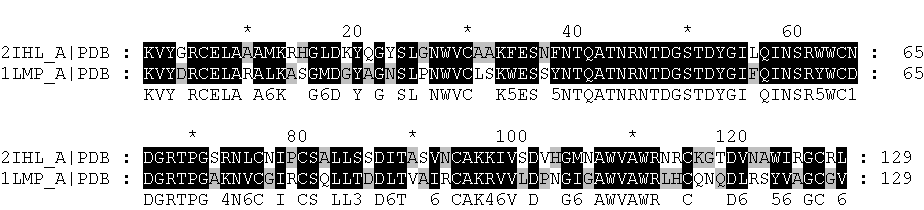
Модификация файла выравнивания:
Переименуем последовательность в файле выравнивания:
После имени последовательности моделируемого белка добавлена строчка:Было Стало >>P1;2IHL_A|PDBID|CHAIN|SEQUENCE >P1;2ihl >P1;1LMP_A|PDBID|CHAIN|SEQUENCE >P1;1lmp sequence:ХХХХХ::::::: 0.00: 0.00
эта строчка описывает входные параметры последовательности для modeller.
После имени последовательности белка-образца добавлено:structureX:1lmp_now.ent:1 :A: 132 :A:undefined:undefined:-1.00:-1.00
эта строчка описывает, какой файл содержит структуру белка с этой последовательностью, номера первой и последней аминокислот в структуре, идентификатор цепи и т.д. В конце каждой последовательности добавлены символы/.
Символ "/" означает конец цепи белка. Точка указывает на то, что имеется один лиганд (если бы было два лиганда стояли бы две точки).
модифицированное выравнивание
Модификация файла со структурой:
удалим всю воду из структуры (в текстовом редакторе)
всем атомам лиганда присвоим один и тот же номер "остатка" (MODELLER считает, что один лиганд = один остаток) и модифицируем имена атомов каждого остатка, добавив в конец буквы A, B, C. Смысл операции в том, что атомы остатка 130 имели индекс А, атомы остатка 131 имели индекс В и т.д. . После модификации имен атомов измените номера остатков на 130.
Итоговый файл:
1lmp_now.ent
Создание управляющего скрипта lysc_cotja.py
В скрипте указано:
- что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4)
что дополнительно нужно сохранять взаимное расположение определенных пар атомов (3.5 ангстрема);
В данном случае трех атомов белка, образующих водородные связи с тремя атомами лиганда - строчки 5-7 с ID пар атомов; параметры взаимного расположения атомов пары описаны в строчке 9-10. 3 точки могут однозначно расположить сложную структуру в пространстве, поэтому мы выбираем водородные связи как источник данных точек.
- что ковалентные связи в гетероатомах нужно вычислять по расстояниям между атомами (так же, как это делает Rasmol), строчка 12
- имя файла с выравниванием и имена последовательностей образца и моделируемого белка, строчка 13 (а имя файла со структурой содержится в выравнивании)
- число и номера моделей, которые нужно построить (в данном примере 5 моделей), строки 14-15
- что пора строить модель строчка 16
- что нужно использовать стандартные валентные углы в полипептидной цепи (строчка 4)
Полученные с помощью скрипта модели.
Качество моделей по WHATIF:
Модель Anomalous bond lengths
RMS Z-score,
RMS-deviationRamachandran plot evaluation
Z-scoreAnomalous bond angles
RMS Z-score,
RMS-deviation2ihl_1.pdb 0.917,
0.0180.079 1.270,
2.1942ihl_2.pdb 0.919,
0.018-0.085 1.358,
2.3552ihl_3.pdb 0.909,
0.0180.512 1.282,
2.2472ihl_4.pdb 0.920,
0.0180.477 1.297,
2.2332ihl_5.pdb 0.904,
0.018-0.072 1.303,
2.290Лучшая
модель:2ihl_5 2ihl_5 2ihl_1
Никаких серьезных отклонений в структурах нет. лучшей можно признать 5-ую структуру.
Картинки из PyMOL c наложенными структурами полученных моделей: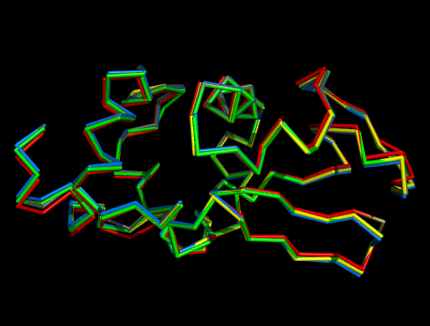
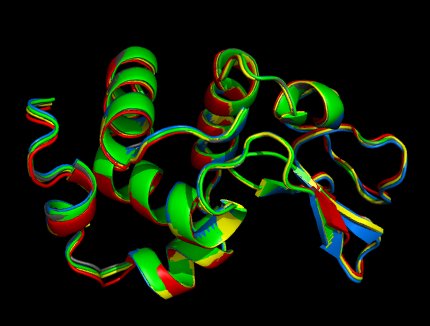
Видно, что структуры очень похожи друг на друга, кардинальных различий нет. Отличаются они в основном в петлях.