- Даны файлы:
- Координаты пептида, 2xl1.pdb.
- Файл с ячейкой уравновешеных молекул формамида, fam_em.gro
- Файл дополнительной топологии для формамида, fam.itp.
- Файл праметров для минимизации энергии em.mdp.
- Файл праметров для "утряски" воды pr.mdp.
- Файл праметров для молекулярной динамики md.mdp
- Построим файл топологии системы в силовом поле amber99sb и файл с координатами в формате Gromacs.
pdb2gmx -f 2xl1.pdb -o pep -p pep -ff amber99sb -water tip3p
Сделаем небольшой отступ в ячейке от ДНК.
editconf -f pep.gro -o pep_ec -d 1.5
Проведём оптимизацию геометрии системы, что бы удалить "плохие" контакты в молекуле.
grompp -f em -c pep_ec -p pep -o pep_em -maxwarn 1
mdrun -deffnm pep_em -v
Изменение максимальной силы в ходе оптимизации геометрии максимальная сила уменьшается (почти каждый раз на новом атоме), падая в итоге на 1 порядок: в начале
F-max = 4.37039e+03 (atom 146), в конце Maximum force = 3.2095328e+02 (atom 30). Оптимизировать о значения Fmax <1, как видно, не получилось.
- Добавим в ячейку молекулы формамида. Количество добавленных молекул формамида - 902.
genbox -cp pep_em -p pep -cs fam_em.gro -o pep_s
- Теперь надо изменить в текстовом редакторе файл тополгии pep.top. После строчки:
; Include forcefield parameters
добавим #include "fam.itp"
; Include forcefield parameters
#include "fam.itp"
Добавим количество молекул формамида в запись [ molecules ]
1
стало:
[ molecules ]
; Compound #mols
Protein 1
FAM 902
- Нейтрализуем заряд системы. Это делаем в два шага: строим tpr и запускаем genion. В выводе grompp сказано, что заряд системы -1,
т. е. необходим 1 положительный ион для нейтрализации заряда системы (-np 1).
grompp -f em -p pep -c pep_s -maxwarn 1 -o pep_s
genion -s pep_s -o pep_si -p pep -np 1
- Проведём "утряску" формамида:
grompp -f pr -c pep_si -p pep -o pep_pr -maxwarn 1
mdrun -deffnm pep_pr -v
Переформатируем pep_pr.gro и pep_si.gro в pdb формат.
editconf -f pep_pr.gro -o pep_pr.pdb
editconf -f pep_si.gro -o pep_si.pdb
Сравним визуально в PyMol изменеия в системах
 |
 |
| Изначально молекулы растворителя четко упорядочены. |
После утряски молекулы растворителя располагаются более хаотично. |
- Скопируем файлы на суперкомпьтер.
Запускаем тестовое моделирование на суперкомпьтере.
grompp -f md -c pep_pr -p pep -o pep_md -maxwarn 1
mpirun -np 16 -maxtime 5 -q test /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Можно записать номер задачи и проверить ход счета в файле mdrun_mpi.out-....
При отсутствии ошибок переходим к основному моделированию.
Запускаем основное моделирование на суперкомпьтере.
mpirun -np 16 -maxtime 1200 /home/golovin/progs/bin/mdrun_mpi -deffnm pep_md -v
Анализ результатов.
Параметры моделирования
- Силовое поле используемое при построении топологии: amber99sb.
- Заряд системы: -1.
- Размер и форма ячейки: параллелепипед со сторонами (нм) 5.06500, 4.67000, 4.22100.
- Минимизация энергии:
- Алогритм минимизации энергии: l-bfgs
- Алгоритм расчёта электростатики и Ван-дер-Ваальсовых взаимодействий:
Coulomb cut-off; LJ or Bcukingham cut-off
- Модель, которой описывался растворитель: FAM (формальдегид)
- Утряска растворителя:
- Параметр который обуславливает неподвижность биополимера: define = -DPOSRES
- Число шагов: 104.
- Длина шага: 1 фс.
- Алгоритм расчёта электростатики - pme, Ван-дер-Ваальсовых взаимодействий - Cut-off.
- Алгоритм термостата Berendsen, баростата - нет.
- Основной расчёт МД:
- Время моделирования: 7 часов 12 минут 42 секунды
количество процессоров: 16
эффективность масштабирования: 100%.
- Длина траектории: 20 нс.
- Число шагов: 10^7.
- Длина шага: 2 фс.
- Алгоритм интегратора: md
(Leap-frog).
- Алгоритм расчёта электростатики - pme, Ван-дер-Ваальсовых взаимодействий - Cut-off.
- Алгоритмы термостата v-rescale, баростата - Berendsen.
- Визуальный анализ движений молекул.
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_pbc_1.pdb -skip 20 -pbc mol
Молекула все время перемещается по экрану, наблюдать за ней не удобно, поэтому применена следующая команда:
trjconv -f pep_md.xtc -s pep_md.tpr -o pep_fit_1.pdb -skip 20 -fit rot+trans
Наблюдения, которые можно сделать из полученного ролика:
№ модели,
время | Описание | Изображение |
| 0, t=0 | Изначально пептид - альфа-спираль
с 4-мя оборотами. | 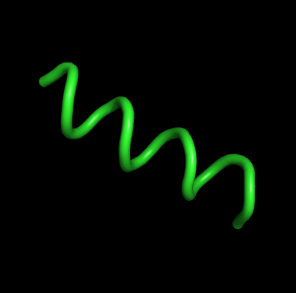 |
| 5, t=1000 | Начинает плавиться N-конец. |  |
| 13, t=2600 | Начинает плавиться C-конец. |  |
| 33, t=6600 | 30-70 модели - наиболее расплавленные,
количество витков 2-3 | 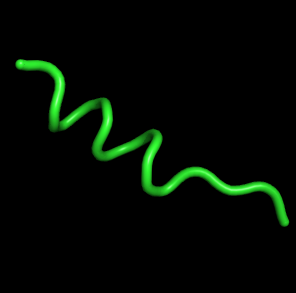 |
| 58, t=11600 | 30-70 модели - наиболее расплавленные,
количество витков 2-3 |  |
| 72, t=14400 | Пептид начинает обратно закручиваться
в спираль. | 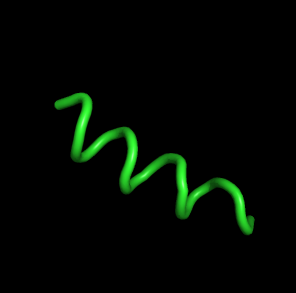 |
| 100, t=20000 | В конце моделирования пептид снова
похож на спираль, хотя и не такую закрученную,
как изначально (около 3-х витков). |  |
- Определение средне-квадратичного отколнения в ходе моделирования.
Так как у нас происходит конформационный переход, сначала рассчитаем отклонение
в ходе всей симуляции относительно стартовой структуры.
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_1
- отклонение постепенно увеличивается примерно до 0.4, затем снова уменьшается до 0.2-0.3.
И относительно каждой предидущей структуры на растоянии 400 кадров.
Если ближе к концу закончился конформационный переход, то отколнение должно уменьшаться.
g_rms -f pep_md.xtc -s pep_md.tpr -o rms_2 -prev 400
- отклонение постепенно увеличивается примерно до 0.5, затем уменьшается и колеблется 0.2-0.3-0.4. (0.3 в конце)
- Определение изменение гидрофобной и гидрофильной поверхности в ходе конформационного перехода.
g_sas -f pep_md.xtc -s pep_md.tpr -o sas_pep.xvg
Зависимость изменения гидрофобной гидрофильной поверхностей доступных
растворителю от времени (красный - гидрофобная, зеленый - гидрофильная):
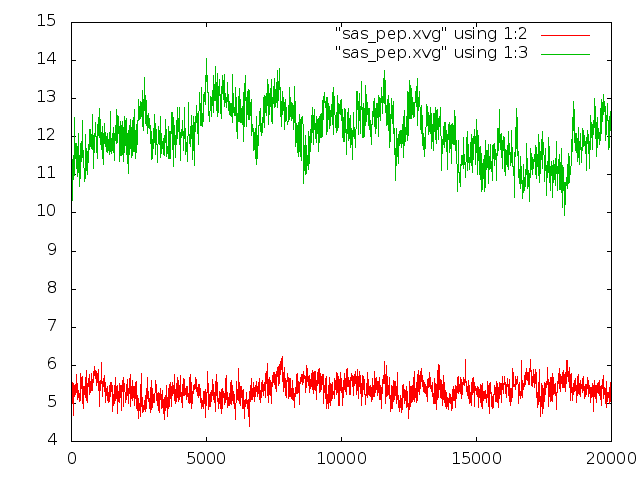
На ролике было видно, что наиболее расплавлен пептид в середине моделирования, когда, согласно графику, гидрофильная поверхность, доступная растворителю, максимальна.
Но изменения в доступности гидрофильной поверхности не слишком велики, для гидрофобной их почти нет.
- Расчёт колчества образуемых водородных связей.
Если мы будем исследовать связи между пептидом и пептидом, то это будут водородные связи в пептиде. Для конца траектории:
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep
Не менее интересно будет изучить количество вдородных связей пептид-формамид.
g_hbond -f pep_md.xtc -s pep_md.tpr -num hbond_pep_sl
График зависимости (красный - в пептиде, зеленый - пептид-формамид):
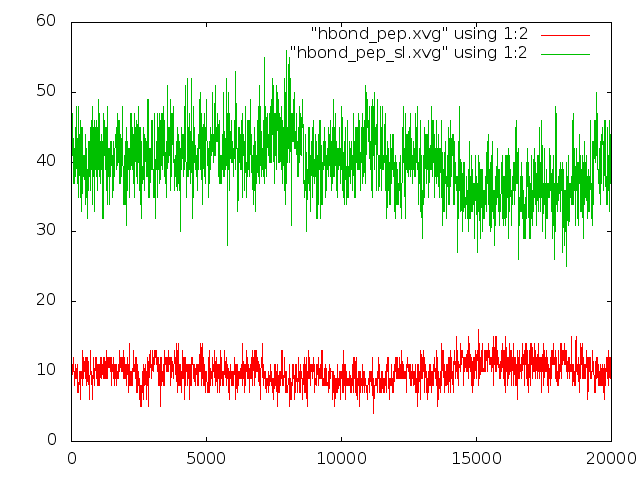
Количество водородных связей в пептиде почему-то почти не меняется, хотя на ролике было явно видно разрушение витков спирали при плавлении.
Количество водородных связей с растворителем несколько варьирует в течение моделирования, но картинка, опять же, не коррелирует с увиденным в ролике.
- Зависимость вторичной структуры от времени:
do_dssp -f pep_md.xtc -s pep_md.tpr -o ss
# Для просмотра переведём xpm в eps
xpm2ps -f ss.xpm -o ss.eps -by 10
Изображение:
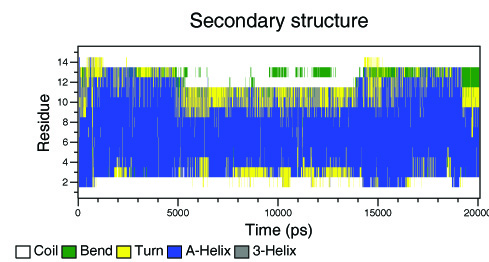
Как и в ролике, на диаграмме видно, что в середине моделирования максимальные концевые участки пептида расплавились из альфа-спирали. Больше всего, как и в ролике, меняется С-конец.