Гомологичное моделирование комплекса белка с лигандом
В этом задании нужно было построить модель выбранного лизоцима (LYS_LAMBD) по последовательности белка и по известной структуре схожего белка (лизоцима форели PDB:1lmp).
import sys
sys.path.append('/usr/lib/modeller9v7/modlib/')
sys.path.append('/usr/lib/modeller9v7/lib/x86_64-intel8/python2.5/')
import modeller
import _modeller
import modeller.automodel
env=modeller.environ()
env.io.hetatm=True
##известная структура лизоцима форели
%%bash
wget http://www.pdb.org/pdb/files/1lmp.pdb
##последовательность выбранного лизоцима
! wget http://www.uniprot.org/uniprot/LYS_LAMBD.fasta
alignm=modeller.alignment(env)
alignm.append(file='P03706.fasta', align_codes='all',alignment_format='FASTA')
## создадим модель
mdl = modeller.model(env, file='1lmp.pdb', model_segment=('FIRST:'+'A', 'LAST:'+'A'))
## и добавим в выравнивание
alignm.append_model(mdl, atom_files='1lmp.pdb', align_codes='1lmp')
## есть смысл поправить идентификаторы
alignm[0].code = 'LAMBD'
alignm.salign()
alignm.write(file='all_in_one.ali', alignment_format='PIR')
## Выбираем объект для моделирования
s = alignm[0]
pdb = alignm[1]
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='all_in_one.ali', knowns= pdb.code , sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
В файле *B99990001.pdb находится оптмизированная структура. Файл с расширением .ini - pdb исходной структуры нашего лизоцима (неоптимизированной).
##визуализация
import nglview
import ipywidgets
w1 = nglview.show_structure_file('LAMBD.B99990001.pdb')
w1
Рис 1. Рассчитанная модель лизоцима.
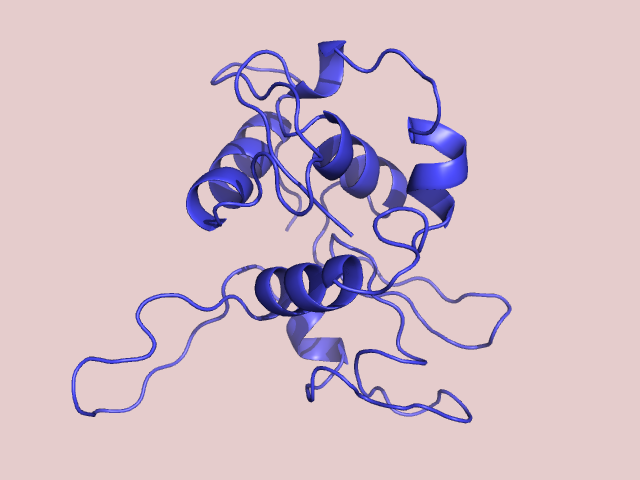
Мы получили модель, однако лиганд был полностью проигнорирован. Чтобы это исправить нужно к последовательности нашего лизоцима добавить "...", что позволит считать три дополнительных остатка лиганда, которые есть в структуре лизоцима форели.
## Получить список остаков
stri = ''
for i in alignm[0].residues:
stri += i.code
stri += '...'
##print stri
## Добавить в объект выравнивание последовательность из строки
alignm.append_sequence(stri)
## Выбираем объект для моделирования
s = alignm[2]
pdb = alignm[1]
s.code = 'LAMBD_L'
alignm.salign()
alignm.write(file='all_in_one.ali', alignment_format='PIR')
print s.code, pdb.code
## Создаем объект automodel
a = modeller.automodel.automodel(env, alnfile='all_in_one.ali', knowns= pdb.code , sequence = s.code )
a.name='mod'+s.code
a.starting_model = 1
a.ending_model = 2
a.make()
import nglview
import ipywidgets
w1 = nglview.show_structure_file('LAMBD_L.B99990001.pdb')
w1
Рис 2. Модель лизоцима с лигандом.

Если мы не уверены точно, как именно расположены лиганд и элементы белка по отношению друг к другу, можно искусственно передвинуть их. В примере ниже мне удается подтянуть свободную петлю белка к лиганду и создать новое взаимодействие. получится новая модель комплекса лизоцима с лигандом.
class mymodel(modeller.automodel.automodel):
def special_restraints(self, aln):
rsr = self.restraints
at = self.atoms
for x,y in [('O7:159', 'N:107')]:
rsr.add(modeller.forms.gaussian(group=modeller.physical.xy_distance,
feature=modeller.features.distance(
at[x],at[y]),mean=3.0, stdev=0.1))
from modeller import *
from modeller.automodel import *
a = mymodel(env, alnfile='all_in_one.ali', knowns= pdb.code , sequence = s.code )
a.make()
import nglview
import ipywidgets
w1 = nglview.show_structure_file('LAMBD_L.B99990001.pdb')
w1
Рис 3. Модель лизоцима со взаимодействием петли и лиганда.
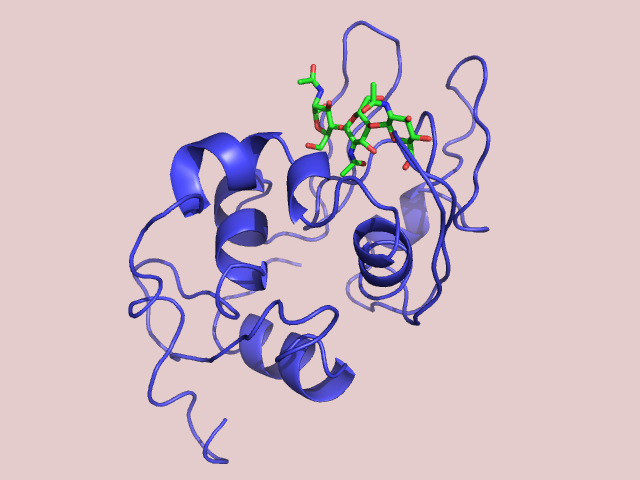
Теперь мы умеем моделировать структуру белка, если уже известна похожая. А также модифицировать результат искусственно.