Множественное выравнивание
Сравнение выравниваний
Для этого задания я взяла белки INS_HUMAN, INS_PIG, INS_CHICH, INS_APLCA.
К сожалению, у меня не получилось написать программу, поэтому я все делала ручками (и глазками), но это было не сложно, так как выбранные мной объекты изучения не длинные и очень схожие между собой.
Я сделала выравнивание в Jalview с помощью Muscle, Mafft и ClustalΩ (получается я сравнила Muscle с Mafft и Muscle с ClustalΩ). Блоки одинакого выровненных колонок: (1+2 программы)(36,84)=(36,84), (114,160)=(122,160); (1+3 программы)(34,40)=(34,40), (48,84)=(48,84), (91,93)=(91,93), (114,160)=(114,160).
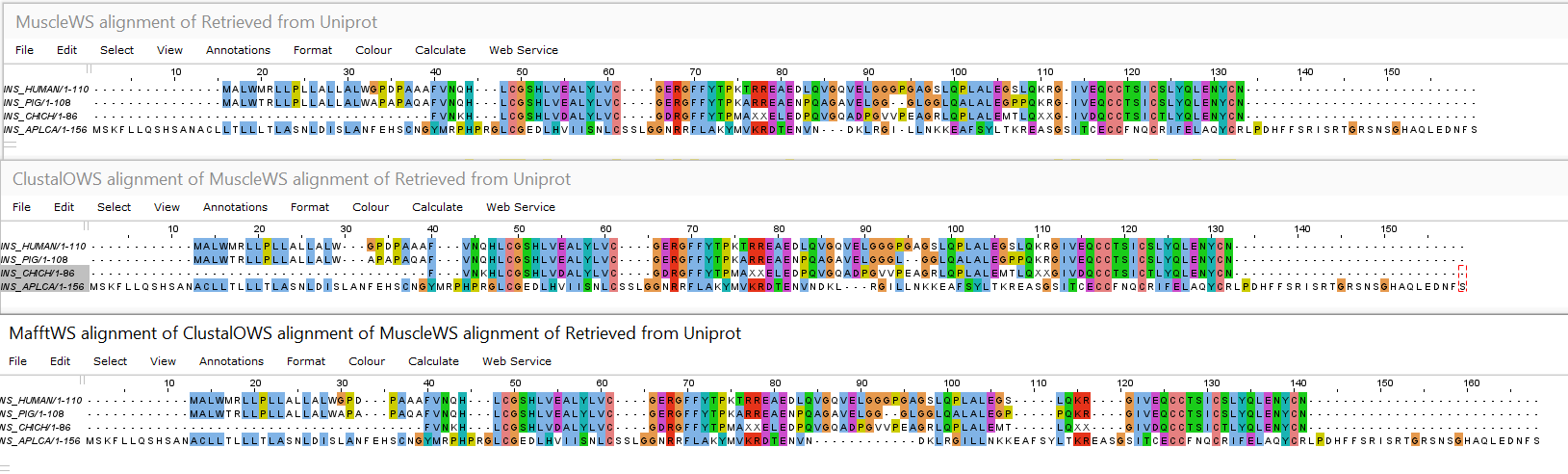
Рис1. Иллюстрация выравниваний.
Получается, что 1 и 3 выравнивания между собой более похожи, чем 1 и 2, так как больше одинаковых блоков. К тому же, в сравненнии 1-3 блоки располагаются на одинаковых позициях (то есть по-разному поставленные гэпы не осуществляют сдвиг последовательности), в то время как в сравнении 1-2 есть разные позиции сходныз блоков (=сдвиг).
Выравнивание 3D-структур
Для оценивания выравнивания 3D структур я взяла белки ACP_ECOLI, ACP_BACSU, ACP_BRUMB (в PDB: 1L0H, 2X2B, 2N57 соответвенно) и загрузила их в PDB, чтобы построить выравнивание по совмещению структур.
На рисунке 2 видно, что пространственные структуры практически полностью совпадают. Даже петли. Единственные места различий - это начало и конец (так как один из белков короче). Это говорит о их высокой схожести.
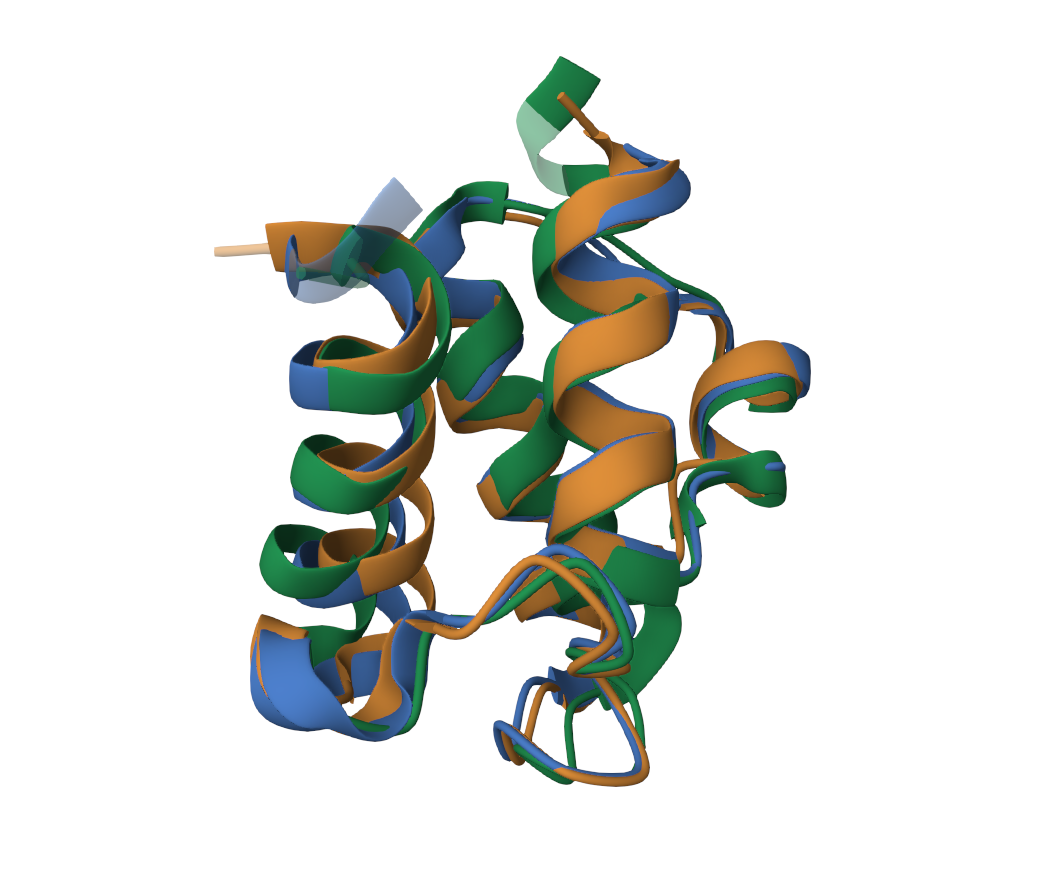
Рис2. Выравнивание вторичных структур (зеленый - ACP_BRUMB, оранжевый - ACP_ECOLI, синий - ACP_BACSU).
Я сравнила выравнивание в Jalview из PDB c выравниванием программой MAFFT. Они очень похожи (кроме концовки). Значит можно сказать, что выравнивание программой отражает в данном случае структурное выравнивание.
Описание программы Clustal Omega.
Clustal Omega (ClustalΩ) - инструмент для множественного выравнивания последовательностей. Она основана на выравнивании последовательностей от самых похожих до самых непохожих, тем самым строя глобальное выравнивание. Эта программа позволяет выровнять очень большое количество последовательностей ДНК/РНК/аминокислот.
Примерная суть действий программы:
Попарное выравнивание по методу mBed. (По истечению попарного выравнивания можно наблюдать выстроенное дерево, которое отражает лишь последовательность выбора цепей, а не ход эволюции). Далее идет группировка последовательностей и снова их соединение, построение более правильного дерева. Получается, что строится дерево от краев (=листьев) к корню.
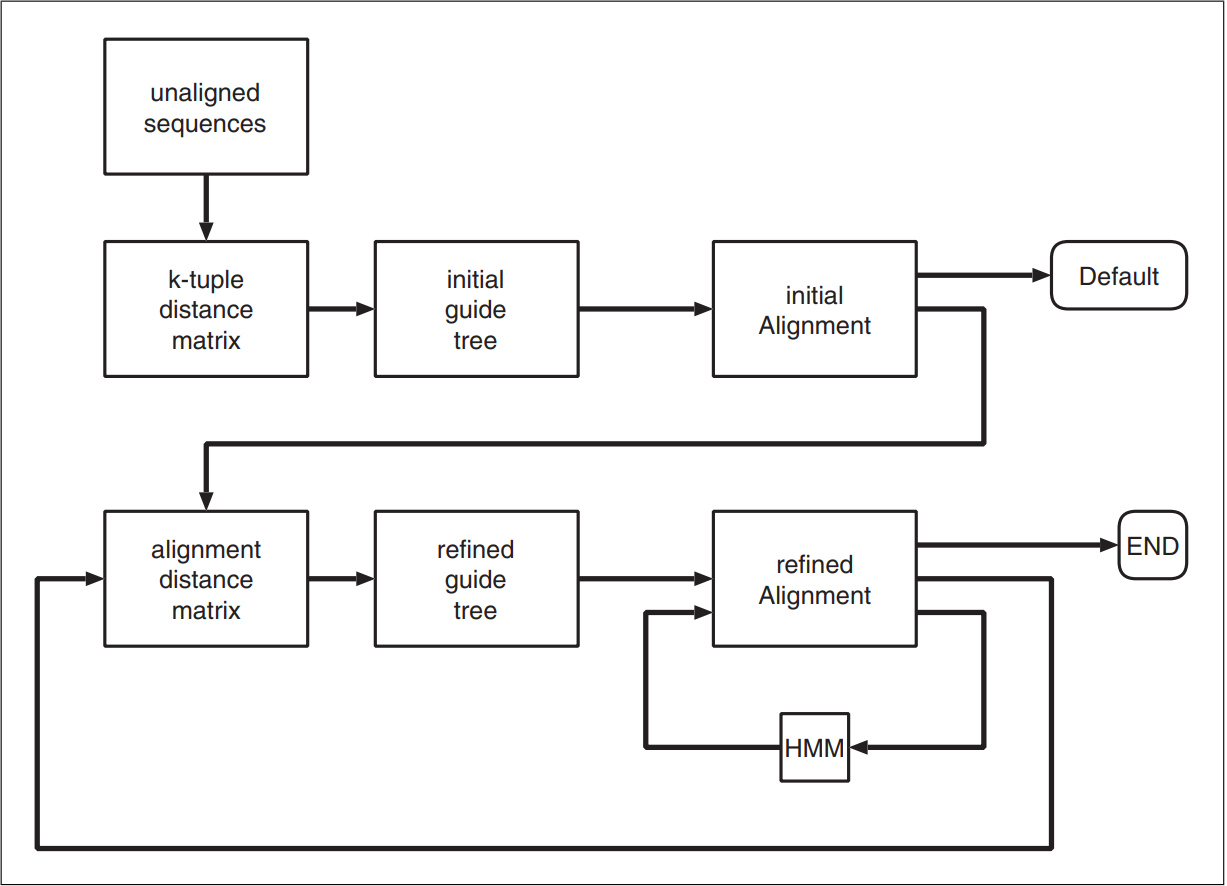
Рис3. Схема из статьи, описывающая алгориитм.
Одна из особенностей данного инстумента - это построение прогрессивных выравниваний, то есть поставленные гэпы и буквы на оппределенные места не меняют своё положение при дальнейшем ходе программы (то есть однажды поставленный знак на какое-то место там и остается до конца, "Часть корабля, часть команды"), а так же построение деревьев в ходе своей работы.
В коммандной строке программа принимает на вход данные в fasta формате (вроде, возможны и другие), строит выравнивание и выдает результат в этом же формате. Важно то, что для построения выранивания необходимы как минимум 3 последовательности.
Другие программы MSA, действующие схожим образом, делают это медленнее и используют больше оперативной памяти, также их точность в множественном выравнивании большого количества последовательностей оставляет желать лучшего, в сравнении с этой программой.