Алгоритмы выравниваний
Выравнивания гомологичных и негомологичных белков
Сравнение выравниваний гомологичных и негомологичных белков
Были скачаны таблицы идентификаторов и длин белков Escherichia coli strain K12 и Bacillus subtilis strain 168. В этих таблицах были найдены пять пар гомологичных белков (с одинаковой мнемоникой функции) и пять пар негомологичных белков с различной мнемоникой функции. Результат объединения таблиц, по которому отбирались белки, можно увидеть по ссылке. Результаты выравниваний представлены в Таблице 1.
Множественное выравнивание
Для построения множественного выравнивания я выбрала белки с мнемоникой функции 6PGD_*, декарбоксилирующую 6-фосфоглюконат дегидрогеназу, и семь модельных организмов: человека, крысу, дрозофилу, E.coli, BACSU, Tripanosoma brucei, Schizosaccharomyces pombe. Было построено выравнивание для семи белков, из которых были выбраны две последовательности, принадлежащие Tripanosoma brucei и Schizosaccharomyces pombe. По результатам выравнивания можно сделать предположение о том, что эти белки, хотя и принадлежат весьма далёким друг от друга организмам, являются гомологичными, так как у них высокий процент сходства и идентичности, малое процентное содержание гэпов. Множественное выравнивание, построенное программой muscle при помощи JalView, в формате проекта Jalview лежит в директории ~term2/block3/credits/Fokina_6pgd.jvp. Скачать проект можно по ссылке. Когда из множественного варианта были удалены все последовательности, кроме двух выбранных, полученное выравнивание я сравнила с выравниванием тех же двух последовательностей, построенных программами needle и water из пакета EMBOSS. На рисунке изображено сравнение трёх выравниваний, верхнее получено из множественного выравнивания (программа muscle), под ним локальное выравнивание, для получения которого использовалась утилита water, нижнее - глобальное выравнивание, полученное при помощи утилиты needle. Важное замечание: для двух нижних выравниваний верхняя последовательность принадлежит дрожжам, а нижняя - трипаносоме, тогда как для верхнего выравнивания всё наоборот, последовательности расположены в обратном порядке. Различия в выравниваниях между различными программами:

Программа water начинает выравнивание с первых кластеризуемых совпадающих позиций, обрезая концевые несовпадающие участки. Две другие программы эти участки включают в выравнивание, причём программа needle выравнивает их между собой несколько иначе, чем muscle, что, вероятно, связано с тем, что в muscle они выровняны ещё и относительно пяти других последовательностей. На первом рисунке видно, что первые два аминокислотных остатка needle выравнивает, а последовательность после них заполняет четырьмя гэпами, а muscle не выравнивает и считает, что первая последовательность короче на четыре остатка.

На втором изображении видно, что программы water и needle выравнивают участки между 304 и 313 аминокислотами одинаково, считается заполненной гэпами, а потом выравнивает между собой 314, 317, 320, 321 и последующие аминокислоты (номера указаны по выравниванию needle), а muscle вставляет гэпы с 14 по 21 аминокислоты, выравнивает 311-314 остатки и вставляет гэп на месте 326 остатка в последовательности белка дрожжей. Следующее различие: в двух одиночных выравниваниях гэп на 332 позиции, а последующие аминокислоты до 349 выровнены, тогда как во множественном выравнивании гэпа на этом месте три и следующие аминокислоты выровнены иначе и выровненных пар меньше. Последнее отличие, заметное на этом рисунке - наличие гэпов с 344 по 347 позиции, которых нет во множественном выравнивании.
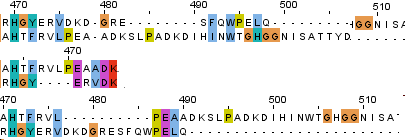
В 479 позиции muscle вставляет гэп (видимо, в других последовательностях этого выравнивания на этом месте была аминокислота),
он также вставляет гэпы с 483 по 490 позиции в белке дрожжей, тогда как needle на этом участке выравнивает сразу три
остатка и гэпы вставляет начиная с 490 позиции. Water концевые несовпадающие участки обрезает, но можно заметить, что участок
с 466 по 470 аминокислоты он заполнил гэпами (для других выравниваний это позиция 474).
На примере этих трёх выравниваний можно заметить, что множественное выравнивание сильно отличается от двух других,
вероятно потому, что в нём участвуют пять других полипептидных цепей. Алгоритмы water & needle сильно схожи сами по себе
и используют одну матрицу замен, поэтому неудивительно, что и выравнивания с их помощью получаются похожими.
Описание ориентированного графа оптимального глобального выравнивания с аффинными штрафами за индели
Оптимальное выравнивание - то, которое имеет больший вес. Вес выравнивания складывается из суммы весов колонок и штрафов за гэпы и индели. Самый большой плюс к весу даёт совпадение аминокислот в колонке, меньший плюс даёт сходство аминокислотных остатков, непохожие аминокислоты дают штраф к весу (вес замены определяется по матрице замен BLOSUM62), буква против гэпа тоже даёт штраф, каждый гэп в инделе даёт штраф, меньший, чем первый гэп в последовательности. Если для построения глобального выравнивания мы берём стандартные параметры, gap opening penalty 10, gap extension penalty 0.5, каждый первый гэп штрафует вес на 10 баллов, каждый следующий гэп, продлевающий индель, даёт штраф в 0.5 баллов. Диагонали на графе указывают на совпадение или несовпадение букв, горизонтальные линии (ось x) - гэпы в первой последовательности, а вертикальные линии - гэпы во второй последовательности. По диагонали мы сдвигаем граф, если в обеих последовательностях в этой колонке стоят аминокислоты, при этом вес ребра берём из используемой матрицы аминокислотных замен. Открытие инделя сдвигает нас по оси z, граф идёт дальше по направлении оси, которая обозначает последовательность, в которой встретился гэп, первый гэп штрафуется теми же 0.5 баллами, считаем, что штраф за открытие гэпа - вес сдвига по оси z, а первый гэп - уже другое ребро. Продолжение инделя отмечается на той же оси, только штраф становится меньше. Вес закрытия инделя равен нулю, при этом граф возвращается по оси z обратно на плоскость x-y, где рёбра идут по диагонали и их вес равен значению в матрице замен для этих двух аминокислот.
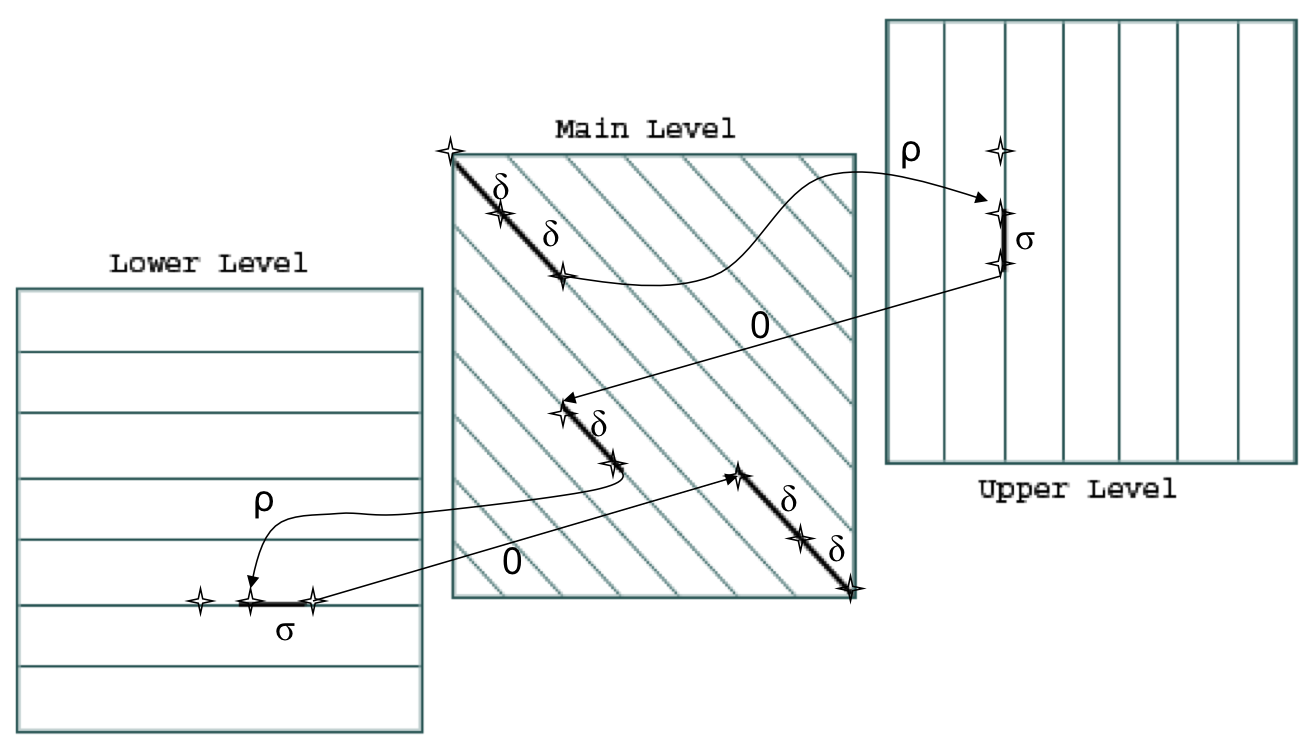
Изображение взято из источника. Main level - плоскость, на которой граф располагается в случае, когда в двух последовательностях в этой колонке стоят буквы, delta - вес ребра, который смотрим в матрице замен. Upper level - плоскость, в которую граф переходит, когда гэп встречается в последовательности, соответствующей вертикальной оси. P - штраф за переход на эту плоскость, gap opening penalty. Sigma - gap extension penalty, штраф за каждый гэп инделя. Lower level соответствует гэпам в последовательности, записанной на горизонтальной оси.
Описание ориентированного графа для построения оптимального ЛОКАЛЬНОГО выравнивания с линейными штрафами за гэпы
Алгоритм аффинного штрафования гэпов основан на том, что более вероятно вырезание или вставка сразу нескольких аминокислот, то есть целого участка-инделя, чем одного остатка. Линейные же штрафы за гэпы этого не учитывают, и штраф за индель прямо пропорционален числу символов гэпа, то есть равен весу одного гэпа умноженному на количество гэпов в инделе. Идея локального выравнивания в том, что выравниваются не обязательно целиком обе последовательности, возможно выравнивание отдельных их частей, что, возможно, позволяет обнаружить гомологичные участки (домены) в белках, которые могут сильно отличаться. Соответственно, концевые гэпы в локальном выравнивании не штрафуются и программы вроде water для построения локальных выравниваний могут их обрезать, за счёт чего выравнивание покрывает не всю последовательность, и мы можем понять, какая часть последовательностей покрывается выравниванием, и на основании этого возможно сделать какие-то выводы об их сходстве. Для построения этого графа мы будем применять ту же матрицу, только в ней все отрицательные веса замен превратятся в нули. Благодаря такой модификации матрицы программа может начать выравнивание из любой вершины графа и в любой же вершине его закончить, игнорируя при этом "неудачные" участки, обладающие маленьким весом и плохо выравнивающиеся между собой. Алгоритм ищет вершину с наибольшим весом (вес вершины в динамическом программировании определяется как сумма весов ведущих в неё рёбер и вершин, из которых они выходят) и строит граф в обратную сторону от этой вершины, пока не натолкнётся на ноль в значении веса вершины. Индель штрафуется 10 баллами, если мы используем стандартные параметры программы.
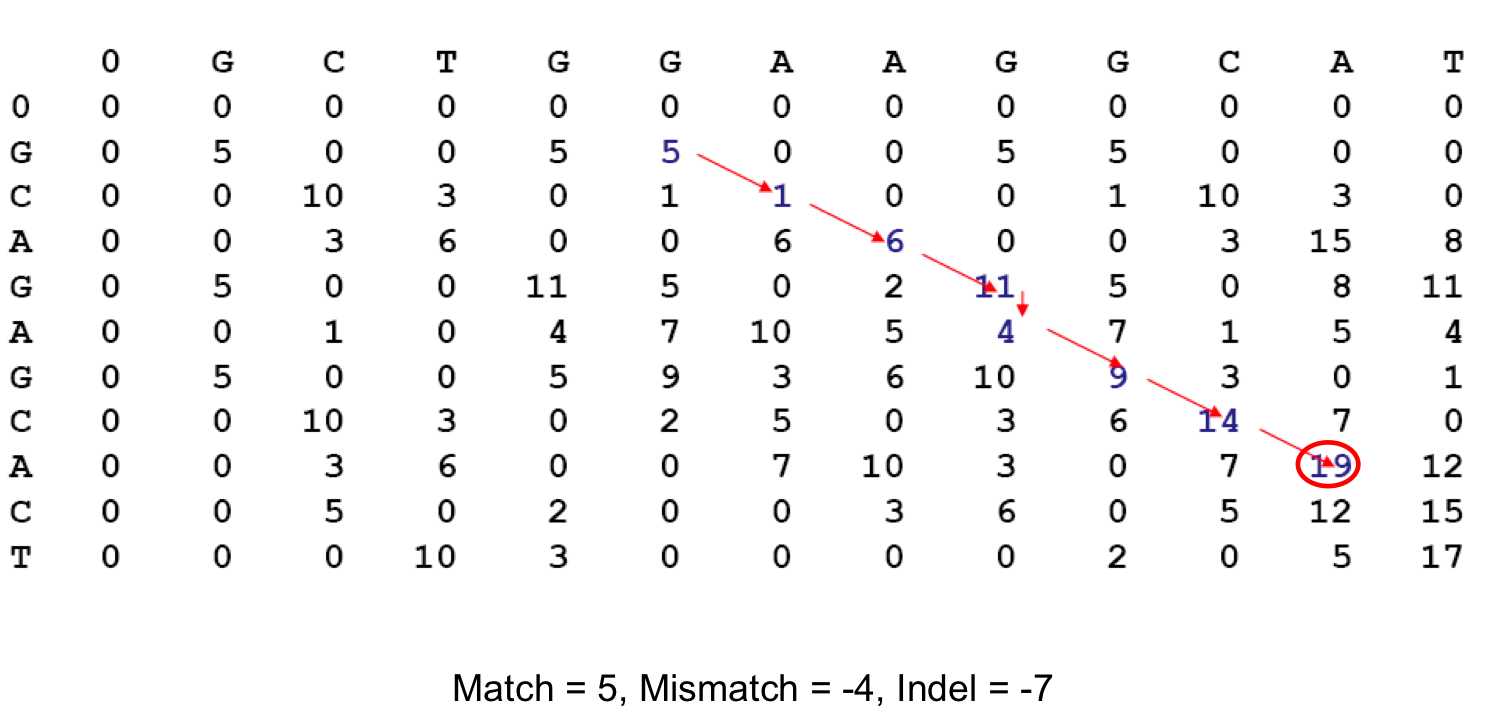
Изображение взято из указанного выше источника, на нём можно увидеть пример графа, описывающего выравнивание двух последовательностей. В вершинах указаны веса замен из матрицы, каждый мэтч (совпадение) даёт +5 баллов к весу выравнивания, несовпадение штрафует 4 баллами, индель отнимает от веса выравнивания 7 баллов, в итоге в конечной вершине получаем 19, это самый большой вес вершины, значит эта вершина действительно последняя в выравнивании.

Итоговый граф выглядит примерно так (изображение из того же источника), его отличие от графа в предыдущем задании в том, что стрелочки идут из первой вершины выравнивания в каждую другую вершину (на рисунке показана лишь часть стрелочек), а из каждой вершины стрелочка идёт к конечной (эти стрелочки вообще не изображены), причём веса всех новых стрелок равны нулю (потому что у нас не может быть отрицательных весов). (Простите за размер картинок я знаю что выглядит ужасно)