Blast
В этом практикуме я познакомилась с работой сервиса Blast, который ищет гомологичные белки или нуклеиновые кислоты по первичной структуре или её фрагменту. В первом задании я изучала возможные параметры поиска на сервере BLAST и по ним находила белки, вероятно гомологичные белку, который я изучала в 5 практикуме. Это ГТФ-уиклогидролаза FolE2, принадлежащая бактерии Nitrosomonas europaeae. Затем при помощи программы JalView из найденных последовательностей я отбирала те, которые являлись гомологичными, и во втором задании строила карту выравнивания для двух белков из этого списка гомологичныъ последовательностей, в последствии анализируя эту карту. Третье задание показало, как можно изменять параметры поиска в этом сервисе и как влияет на результаты изменение параметров.
Поиск гомологов моего белка в базе данных SwissProt
В базе данных SwissProt ID моего белка - GCH4_NITEU. Для нахождения его гомологов в сервисе BLAST я выбрала раздел protein,
ввела в поисковую строку ("Enter Query Sequence") идентификатор (UniProt AC) фермента - Q82VD1. В этом окне можно также ввести
последовательность аминокислот в формате fasta или загрузить файл.
Query subrange - в этом окне можно указать начало и конец участка полипептида, если мы хотим искать не по всей последовательности, а только по её отрывку. Я оставила это окошко пустым.
Align two or more sequences - можно выровнять несколько последовательностей друг относительно друга. Я не поставила здесь галочку.
Database - выбор базы данных, в которой будет искать последовательности алгоритм. Я оставила дефолтное значение Non-redutant protein sequences (nr).
Organism - ограничение поиска по организму, алгоритм выведет только белки, принадлежащие выбранному организму или нескольким. Выбрав Exclude, можно исключить из результатов поиска белки конкретных организмов.
Algorithm - используемый алгоритм, я оставила дефолтное значение blastp (protein-protein BLAST).
Max target sequences - максимальное количество выводимых последовательностей, которые выровнялись с нашим белком. Если всего находок больше числа в этом окошке, выводятся те из них, которые при выравнивании набрали наибольшее значение score. Я установила этот параметр на 20000.
Short queries - у BLAST есть специальные параметры для обработки коротких последовательностей, которые применяются, если
в этом окошке поставить галочку. Я галочку не ставила, потому что моя последовательность не короткая.
Expected treshold - задаёт верхний порог E-value, математического ожидания числа находок BLAST с таким же или большим
весом в случайном банке того же размера. Я оставила дефолтное значение этого параметра - 10, то есть белки с E-value выше десяти в выборку не попадут.
Word size - размер индексированных слов-затравок, на которые бьётся последовательность, я оставила изначальное значение - 6.
Max matches in a query range - ограничение числа выравниваний с конкретным участком белка, чтобы большое число совпадений
с этим участком может помешать найти незначительные совпадения с другими участками. Я
Matrix - выбор матрицы замен, по которой алгоритм будет считать вес выравнивания. Я выбрала стандартную матрицу BLOSUM62.
Gap Costs - штрав за первый гэп (Existence) и за продолжение инделя (Extension). Я оставила дефолтные параметы 11 и 1.
Compositional adjustments - выбор способа борьбы с участками малой сложности, которые могут быть
сильно схожи, но это сходство биологически бессмысленно. Я поставила Conditional compositional score matrix adjustment.
Filter Low complexity regions - включает маскировку участков малой сложности. Я не поставила здесь галочку.
Mask for lookup table only - включает маскировку участков малой сложности на первом этапе работы алгоритма, то есть при составлении таблицы. Я не поставила галочку в этом окошке.
Mask lower case letters - маскирует строчные буквы во введённой последовательности. Я не поставила здесь галочку.
Последовательности в выдаче вообще имеют довольно высокую степень гомологии (так, на цветовой схеме выравнивания
большинство красных линий одной длины или чуть короче по краям, то есть покрытие выравнивания весьма велико). В выданных последовательностях были выбраны 9 с разными значениями E-value и разной степенью гомологии. Найти белки с другими названиями
у меня не получилось, но некоторые белки из списка гипотетические, то есть их функция не установлена, и я решила, что будет интересно выровнять их с ГТФ-циклогидролазами, чтобы проанализировать их на гомологию.
Эти девять белков можно скачать по ссылке
А вот тут ссылка на табличку с результатами.
Я построила выравнивание при помощи алгоритма Muscle в Jalview с дефолтными параметрами, потом из девяти белков выбрала те четыре,
которые показались мне наиболее гомологичными (у них было больше совпадающих аминокислот и друг с другом они выравнивались почти без гэпов).
Не гомологичные пять последовательностей я удалила, а оставшиеся перевыровняла и покрасила с применением Above Identity treshold 100%.
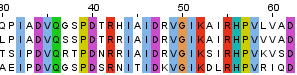
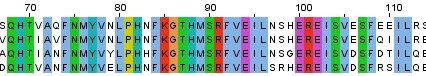
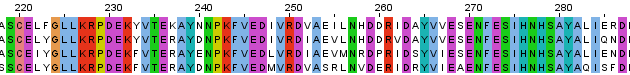
На картинке видно, что для этих последовательностей существует один довольно протяжённый участок, по которому их можно считать гомологичными, с 32 по 287 аминокислоты. Этот участок начинается и заканчивается полностью консервативной позицией и плотность консервативных позиций в нём весьма высока, есть даже участки, например с 84 по 96 аминокислоты, в которых во всех колонках аминокислоты совпадают, колонок с гэпами в нём нет. Проект Jalview с выравниванием этих четырёх белков.
Объясниение карты сходства двух белков
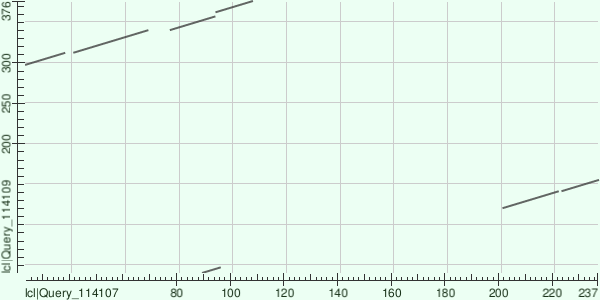
Я выбрала по одному идентификатору из первой (A0A1R4EYK5_9MICO) и второй (A0A0C2WMX0_AMAMU) групп. Карта локального сходства изображена на рисунке. Эта карта демонстрирует участки сходства последовательностей, которые выровнялись при помощи алгоритма BLAST. Алгоритм нашёл три сходных участка в последовательностях двух негомологичных белков разных организмов. Второй и третий выровнянные участки слишком короткие и имеют очень большое значение E-value - 3.2 и 5.3 соответственно, а также незначительный процент совпадающих и сходных аминокислот. Алгоритм выровнял начало первой последовательности (с 24 по 108 аминокислоты) с концом второй (с 298 по 376). Оно изображено в левом верхнем углу карты, разрывы линии соответствуют инделям, появившимся в результате делеции трёх и восьми аминокислот во второй последовательности (разрывы со сдвигом по горизонтали) и пяти в конце первой (маленький разрыв со сдвигом линии по вертикали). Чем длиннее участок делеции, тем длиннее будет разрыв на линии. Второе выравнивание длиной всего 6 аминокислот, с 90 по 96 для первой и с 41 по 47 для второй последовательности, в нём нет гэпов и инделей, на карте локального сходства оно изображено короткой линией внизу в середине. Третье выравнивание тоже не очень длинное, с 202 по 237 аминокислоты для первой последовательности и со 121 по 155 аминокислоты. Это конец первой последовательности (изображённой по горизонтали) и середина второй, на карте локального сходства это выравнивание изображено линией в правом конце, разрыв на линии соответствует единственному гэпу, появившемуся в результате делеции.
Игры с BLAST
Я взяла последовательность "I KEEP six honest serving-men" и ввела её в строку поиска, оставив все параметры дефолтными и убрав галочку у параметра "automatically adjust parameters for short input sequence". Минимальный E-value в выдаче составил 1.3, что слишком много, а значения для других последовательностей и того больше, что значит, что последовательности нельзя считать гомологичными (неудивительно, ведь я ввела точно не белковую последовательность. Значения Score невелики, что опять же свидетельствует о случайном сходстве а не о гомологии. Зато процент идентичности для некоторых последовательностей доходит до 100%, то есть участок, найденный в последовательности, полностью совпал с участком введённой фразы. На рисунке изображено соотношение найденных последовательностей с введённой, чёрный цвет линий свидетельствует о небольшом значении score, а расположение линий показывает, какой участок последовательности выровнялся с другой последовательностью. Большинство найденных последовательностей выравниваются с серединой введённой фразы, но есть и те, которые выравниваются с ней почти на всю её длину или с концевыми её участками.

Следующее, с чем я "играла", это параметры поиска, которые можно менять и соответственно менять выдаваемые результаты. При смене матрицы с BLOSUM62 на BLOSUM90 ничего не изменилось, выдача осталась такой же. При замене матрицы на PAM30 минимальное значение E-value изменилось на 5е-136 вместо 6е-138. Изменение матрицы аминокислотных замен увеличивает точность поиска - так, матрица PAM260 умеьшает максимальный вес вырваниваний в выдаче (то есть алгоритм находит последовательности с меньшим весом выравнивания и не находит те, которые обладают большим весом и находятся, если использовать другие матрицы замен.) При установлении Gap Costs на existence: 6 Extension: 2 Вес выравнивания с одной и той же последовательностью, принадлежащей Azospira oryzae, падает до 404 вместо 451. При изменении длины слова с 3 на 2 сохраняется, сохраняются первые несколько последовательностей в выдаче и значеня веса и процента идентичности для них. При уменьшении E-value уменьшается объем выдачи, при уменьшении его до значения, меньшего чем минимальное значение этого параметра в выдаче, результатов вообще нет, потому что ни один из них не удовлетворяет параметру. Значение E-value увеличивается для всех последовательностей примерно на 10 порядков (теперь минимальный 5е-126), а вес выравнивания для той же Azospira oryzae становится 338 при установлении параметра Gap Costs на Existence:11 Extension:2. Разницы между алгоритмами PSI-Blast и DELTA-Blast с дефолтными значениями всех параметров не влияет на состав выдачи, хотя принципы работы этих алгоритмов различны, первый основан на выравнивании отдельных элементов, а второй на доменной организации последовательностей. Использование второго алгоритма позволяет проводить поиск в тех случаях, когда мы можем предполагать консервативность доменов и вариабельность междоменных частей, и находить различные белки с разными междоменными участками.