Анализ транскриптомов. Bedtools.
Задание 1. Анализ качества чтений.
В ходе практикума я работала с 4 хромосомой человека, репликой 1.
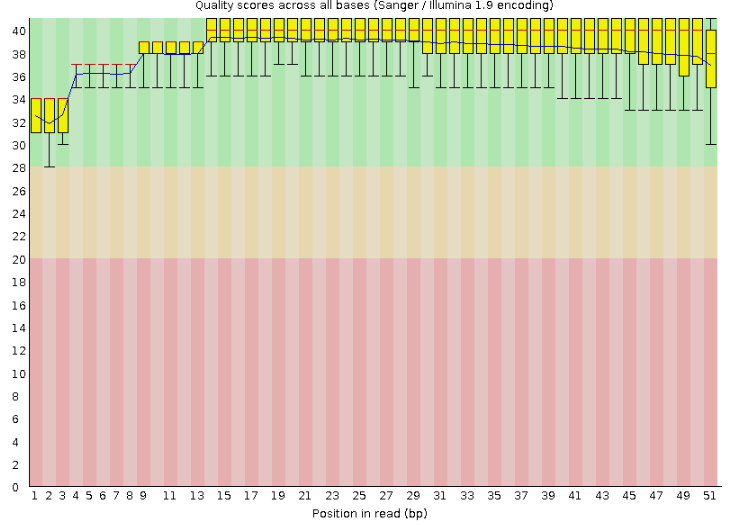
fastqc chr4.1.fastq проверка качества чтений, качество до очистки изображено на первом рисунке.
Как и в предыдущем практикуме, с помощью команды java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr4.1.fastq trimmed.fastq TRAILING:20 MINLEN:50
были очищены чтения с качеством ниже 20 и длиной меньше 50. Изначально чтений было 2735, после чистки осталось 2705 (98,9%),
то есть 30 чтений были удалены.
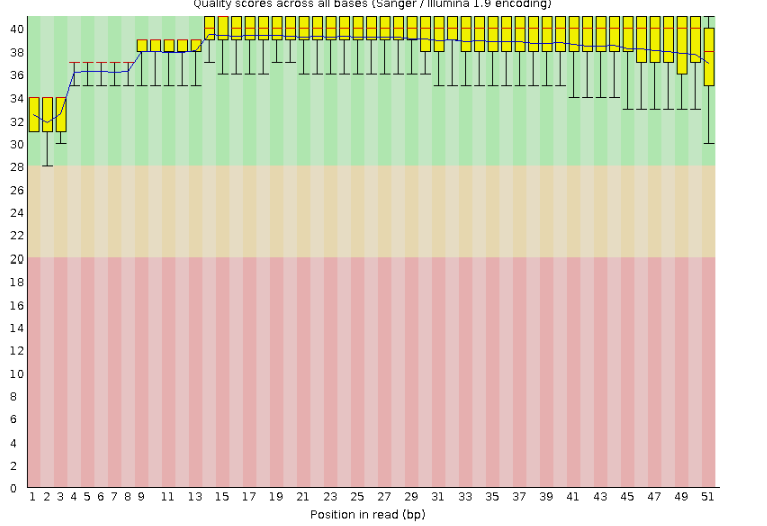
Качество чтений после очистки изображено на рисунке 2. Оно проверено с помощью fastqc trimmed.fastq
После очистки качество чтений не изменилось, видимо, оно изначально было достаточно хорошим (из 1 рисунка видно,
что ни одно из чтений не имеет качества ниже 20).
Задание 2. Картирование чтений.
Для картирования чтений использована команда hisat2 -x chr4 -U trimmed.fastq -S align.sam --no-softclip. По сравнению с запуском этой же команды в предыдущем практикуме, в этот раз я убрала параметр --no-spliced-alignment, потому что здесь на геном картируется транскрипт, который уже прошёл сплайсинг, поэтому смысла в параметре, который заставляет программу картировать чтения без учёта сплайсинга, нет. 72 чтения (2,66%) не откартированы ни разу, 2633 (97,34%) откартированы ровно 1 раз, и ни одного не откартировано больше одного раза. Качество картирования чуть ниже, чем в предыдущем практикуме, но всё ещё достаточно хорошее.
Задание 3. Анализ выравнивания.
samtools view -b align.sam -o align.bam - перевод выравнивания в бинарный формат для упрощения анализа.
samtools sort align.bam sorted - сортировка выровненных ридов по их координатам в референсной последовательности.
samtools index sorted.bam - индексирование отсортированного файла.
Задание 4. Подсчёт чтений.
htseq-count -f bam align.bam -sno -i gene_id -m union gencode.v19.chr_patch_hapl_scaff.annotation.gtf > count.txt - подсчёт чтений. параметр -f задаёт формат входного файла (sam или bam); параметр -s указывает на конкретную цепь, к которой относятся риды (yes, reverse), или же на то, что нужно анализировать их в обоих направлениях (no); параметр -i задаёт индекс или feature ID; параметр -m указывает способ учёта чтений, ложащихся сразу на несколько генов (union если хотим учитывать их все, intrsection-strict если чтение целиком ложится на ген, intersection-nonempty если чение имеет с геном общий участок
Задание 5. Анализ результатов.
grep -wv 0 count.txt > res.txt - создание текстового файла, в котором строчки, не заканчивающиеся нулем (то есть те гены, на которые наложились риды). Всего строчек в файле count.txt больше 63 тысяч, это количество генов на хромосоме, на которую картировалось чтение. Чтения картировались далеко не на все эти гены, файл с результатами выглядит так:
ENSG00000071127.12 1861 ENSG00000223086.1 1 ENSG00000261490.1 2 __no_feature 769 __not_aligned 72Чтения, которые ни на один из генов не легли, могут появиться в результате загрязнения биоматериала чужеродной ДНК, остатками праймеров или поли-А хвостами зрелых матричных РНК. Большинство ридов накладываются на один ген, ещё три - на два других. Есть также довольно много ридов, которые ни на один из генов не наложились. ENSG00000071127.12, на который легло 1861 прочтений, кодирует белок с 9 WD-повторами, которые представляют собой С-концевой домен из 30-40 аминокислот, многие из которых консервативны, в основном состоящий из триптофана и аспарагиновой кислоты. Эти домены участвуют во взаимодействии белков, скорее всего кодируемый этим геном белок помогает запустить разборку актиновых филаментов.
ENSG00000223086.1 - ген, который в базе NCBI не находится. Зато находится в банке Ensembl, является 5S-рибосомальным псевдогеном, кодирует конкретную РНК со сложной вторичной структурой.
ENSG00000261490.1 - в базе NCBI тоже найден не был. В Ensembl он отнесён к типу Sense overlapping - длинным некодирующим транскриптам, содержащимся в интронах кодирующей последовательности. Последовательность ДНК, кодирующая длинную некодирующую РНК с не очень ясными функциями.