ОБработка результатов секвенирования по Сенгеру
Задание 1
В этом задании нужно было проанализировать хроматограмму, данную в двух прочтениях:
Прямое прочтение в формате .ab1
Обратное прочтение в формате .ab1
Для анализа хроматограммы я использовала программу Chromas, т.к. GeneStudio не отображал часть хроматограммы по неизвестной причине. Файлы с прямой и обратной последовательностями были загружены
в отдельные окна программы, затем в файле с обратной последовательностью был применён переход к комплементарной цепочке с помощью функциии Reverse.
Хроматограммы были проанализированы по всей длине, найдены проблематичные нуклеотиды и с помощью выравнивания (ссылка ниже) определены их координаты на второй цепочке,
по которым проверялась правильность автоматического определения нуклеотида. Все исправления в консенсусном файле отмечены маленькими буквами. Выравнивание двух последовательностей проводилось с использованием программы needle:
Выравнивание двух хроматограмм в формате .needle
Консенсусная последовательность, созданная автоматически Gene Studio
Консенсусная последовательность почти не нуждалась в редактуре, кроме замены неопределённых нуклеотидов на конкретную букву из таблицы IUPAC Ambiguity Codes, последовательность
была прочитана хорошо, проблемные нуклеотиды программа нормально исправляла по противоположной цепочке. Даже концевые плохо читаемые концы, обрезаемые Chromas, попали
в консенсусную последовательность, т.к. GeneStudio обрезал сильно меньшей длины участки, а необрезанные участки умудрялся прочитать. По сути редактирование консенсусной
последовательности заключалось в удалении нескольких N на каждом из концов и исправлении оставшихся единичных N на протяжении последовательности. Трудностей и проблемных нуклеотидов в последовательностях почти не было.
Консенсусная последовательность, отредактированная мной
Были найдены участки в начале и конце хроматограмм, имеющие плохое качество прочтения или вообще не читаемые, потом эти участки были найдены автоматически Chromas,
результаты я сравнила и они совпали. Было проанализировано общее качество хроматограмм, оказалось что у обратной последовательности почему-то в среднем ниже уровень шума и
она лучше читаема. Ниже описаны результаты анализа хроматограмм и приведены примеры проблемных нуклеотидов, которые проверялись по второй цепочке.
Для прямой последовательности моё определение нечитаемых концов совпало с автоматическим, начало (5'-конец) нечитаемо до 96 нуклеотида, т.к.
до него сохраняется высокий уровень шума, большое количество идущих подряд неопределённых нуклеотидов и низкое качество определения
для тех, которые всё-таки определены. 3'-конец нечитаем с 677 и до 721 нуклеотида, хотя программа определяет его начиная с 678,
несмотря на то, что 677 нуклеотид неопределён и я бы не стала его включать в хорошо читаемую часть хроматограммы.
Ситуация с нечитаемым концом (5'-конец, который расположен в конце хроматограммы из-за применённой функции reverse) обратной последовательности та же, он начинается
с 620 (621, если верить автоматическому определению)
и заканчивается вместе с концом хроматограммы на 727 нуклеотиде. Нечитаемое начало (3'-конец) Chromas определяет до 30 нуклеотида,
хотя нуклеотиды с 30 по 40 имеют невысокое качество определения, средний уровень шума на этом участке высокий, из 10 нуклеотидов 3 определены как неизвестные (N), так что я бы обрезала начало до 40 нуклеотида.
Интересно, что длины нечитаемых участков для хроматограмм сопоставимы, 5'-нечитаемый участок прямой последовательности имеет длину 96 нуклеотидов, а
3'-конец обратной - 106, 3'-участок прямой последовательности имеет длину 44, а нечитаемый 5'-участок обрытной около 40, что объясняется тем, что обратная цепь юыла сделана обратно комплеменатрной.
Среднее качество хроматограммы для прямой цепочки хорошее, особенно начиная с 310 нуклеотида - после него уровень шума снижается до 20% (определено "на глаз"),
вообще шум не превышает 40% за исключением отдельных нуклеотидов, качество определения тоже довольно высоко, в среднем оно около 70%, хотя для отдельных нуклеотидов падает до 10.
Для обратной цепочки качество прочтения в среднем кажется более высоким, чем для прямой, оно сохраняется выше 60% с 100 до 388 нуклеотида,
потом немного снижается и становится низким (около 30%) с 504 нуклеотида. Уровень шума изменяется примерно так же, как для прямой, но при этом он в среднем ниже. Для прямой
цепи очень сильно отличаются по высоте соседние пики, что не мешает прочтению, но затрудняет его. Обратная цепь в этом плане выглядит лучше, пики, отличающиеся по высоте от
среднего, встречаются реже, и среднюю высоту хотя бы можно определить на протяжении плюс-минус всей хроматограммы.
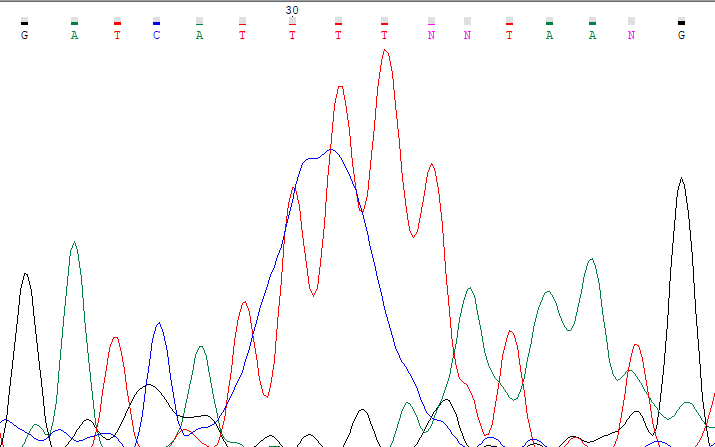
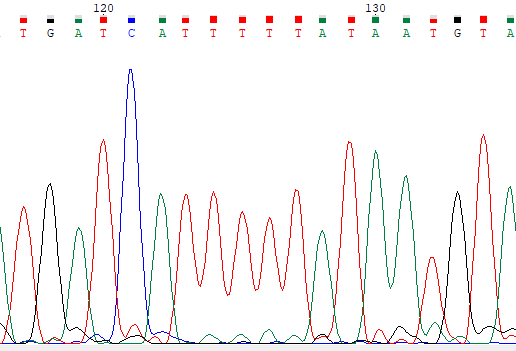
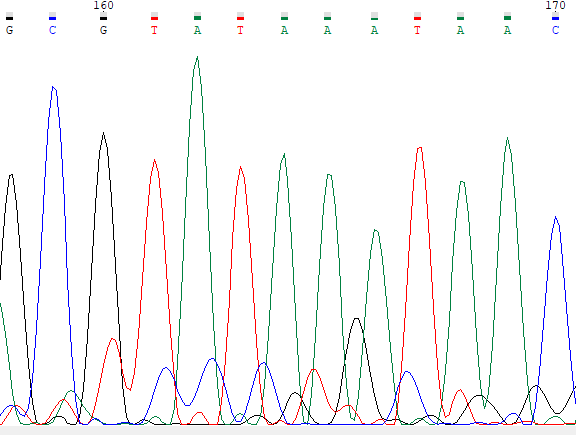
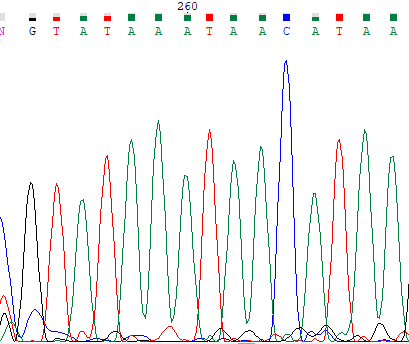
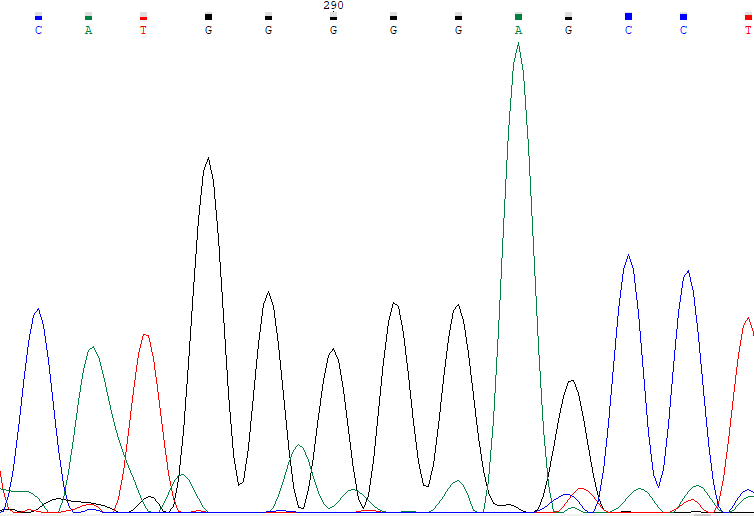
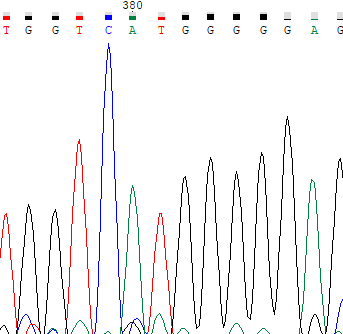
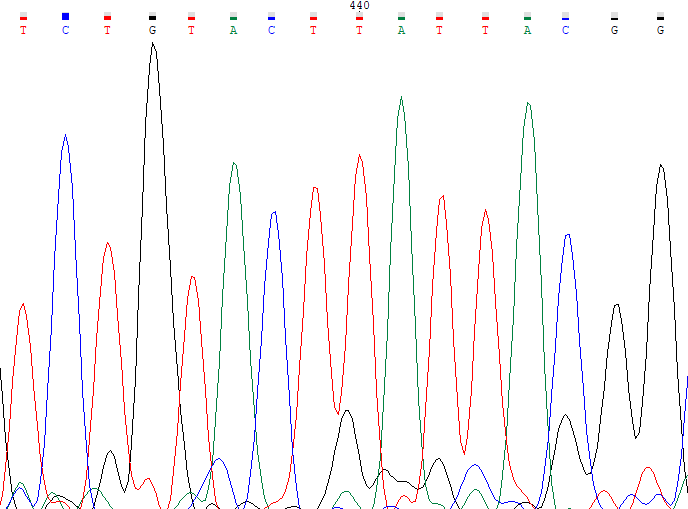
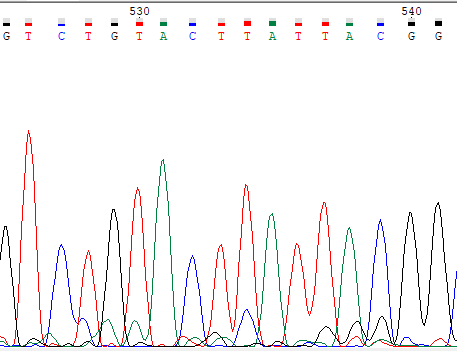
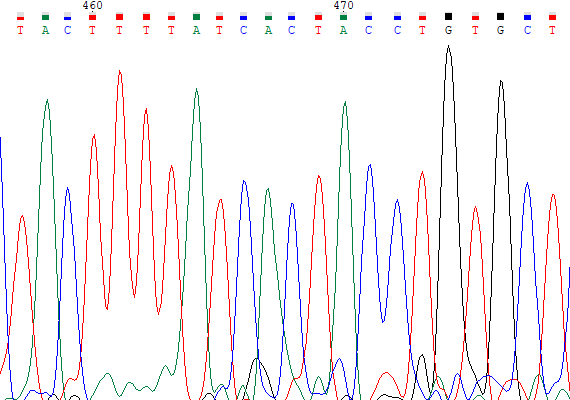
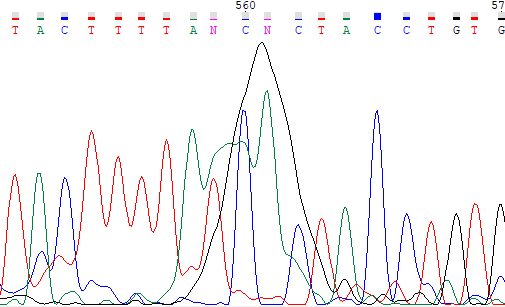
Ниже приведены примеры проблемных нуклеотидов, на левой картинке прямое прочтение, на правой обратное, координаты нуклеотидов
определены с помощью выравнивания.
Задание 2
В задании нужно было привести пример нечитаемой хроматограммы, и для этого я взяла плохо прочитанный участок хроматограммы, с которой я работала в предыдущем задании.
Это начало прямого прочтения, которое имеет низкое качество определения, а многие нуклеотиды не определены вообще.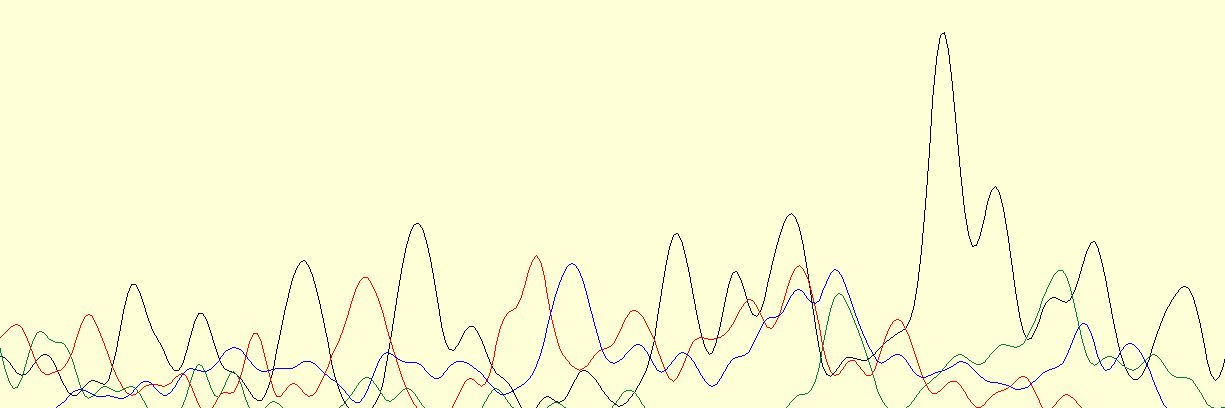
На картинке приведён нечитаемый участок начала хроматограммы. Он нечитаем, т.к. на
всём его протяжении сохраняется довольно высокий уровень шума, пики очень разной высоты и ширины с нечёткими границами, расположены на разном расстоянии друг от друга,
часто пики по высоте сопоставимы с шумом, пики разных цветов и сравнимой высоты накладываются. Невозможно дифференцировать нуклеотид в каждой из позиций,
из-за разной ширины пиков трудно даже установить их количество, один пик может соответствовать больше чем одному нуклеотиду, при этом не будучи разделённым на несколько пиков поуже.
Из-за разной высоты соседних пиков непонятно, какие из них являются шумом, а какие можно считать сигналами, т.к. некоторые
сигналы по высоте совпадают с шумом.