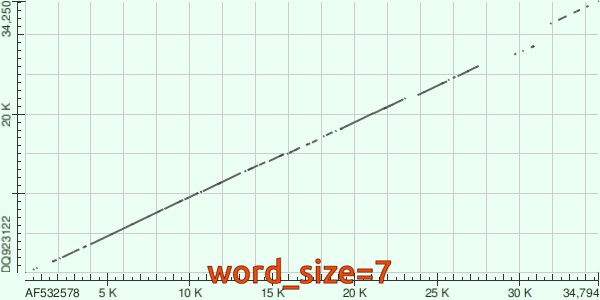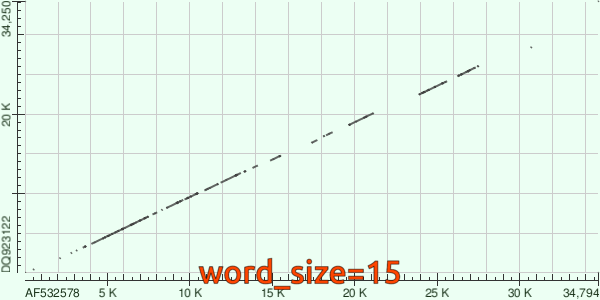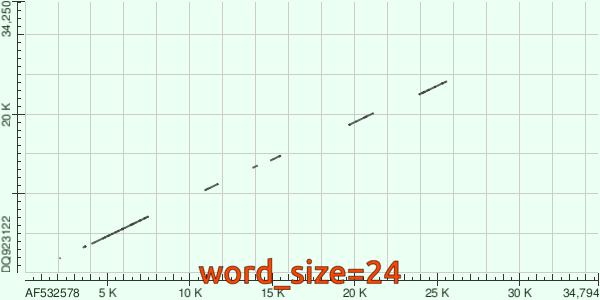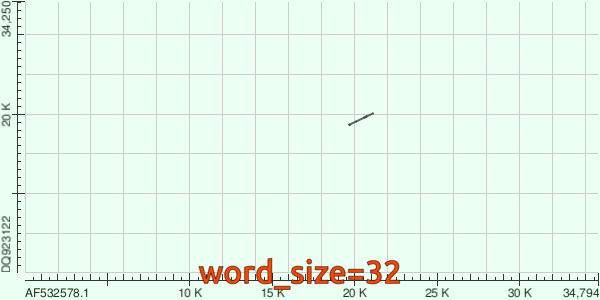# Аденовирусы и их геномы
Сегодня мы поработаем с двумя аденовирусами: 11-м человека и 10-м быка. Будем смотреть на их полные геномы. Названия записей в ENA и запросы, по которым я на них вышел, привожу в виде таблицы:
|Organizm | query |
| ------ | :------: |
| Human adenovirus type 11 strain Slobitski, complete genome | tax_eq(10541) AND description="complete" |
| Bovine adenovirus 10 strain Belfast 1, complete genome | tax_eq(39788) AND description="genome"|
Приведу ещё удобные `curl`-запросы, которые можно выполнить в терминале и получить выдачу, аналогичную той, что на сайте:
```
curl -X POST -H "Content-Type: application/x-www-form-urlencoded" -d 'result=sequence&query=tax_eq(10541)%20AND%20description%3D%22complete%22&fields=accession%2Cdescription%2Ctax_id&format=tsv' "https://www.ebi.ac.uk/ena/portal/api/search"
curl -X POST -H "Content-Type: application/x-www-form-urlencoded" -d 'result=sequence&query=tax_eq(39788)%20AND%20description%3D%22genome%22&fields=accession%2Cdescription%2Ctax_id&format=tsv' "https://www.ebi.ac.uk/ena/portal/api/search"
```
Выбор конкретных аденовирусов я делал на основе филогенетического положения: хотелось бы, чтобы исследуемые организмы были умеренно сближены и умеренно удалены друг от друга.
# megablast и blastn
Основная цель этого пункта, как и всего практикума - увидеть заметные различия в выдаче этих двух алгоритмов. Параметры, с которыми исходно были запущены алгоритмы, привожу в таблице:
| Algorithm| word_size| match | mismatch | gap opening | gap extension |
| ------ | :------: |:------: |:------: |:------: |:------: |
| megablast| 24| 1| -2|linear |linear |
| blastn | 7| 4|-5 |12 | 8|
Итак, посмотрим на `dotplot`'ы, которые были построены по результатам работы каждого из алгоритмов локального выравнивания (первый - megablast, второй - blastn).
Результат выравнивания megablast'ом.
Результат выравнивания blastn'ом.
Мы видим, что megablast установил гомологию только между небольшим количеством участков геномов двух вирусов, тогда как по результатам blastn эти геномы сильно схожи почти во всей длине.
Так происходит потому, что у megablast'а гораздо длиннее "якорь", т. е. для его работы последовательности индексируются более длинными частями (в нашем примере длина составляет 24 nt). Это делает мегу гораздо более чувствительным к мутациям (при этом пусть даже синонимичным, алгоритм нуклеотидный и про это ничего не знает), поэтому логично, что для не слишком филогенетически близких организмов у него будет мало возможностей найти совпадения длиной 24 нуклеотида, чтобы начать с них выравнивание.
В это время blastn с `word_size=7` находит куда больше гомологичных участков, что закономерно: шанс встретить последовательность длиной 7 нуклеотидов, которая не отличается у исследуемой пары организмов намного выше, стало быть, и выравниваний с таких якорей инициируется хотя бы физически *больше*.
Справедливо будет заметить, что описанная картина - не только типичный пример того, что megablast и blastn существуют для несколько разных целей, но и повод подумать про пользу совместного использования этих алгоритмов. Действительно, имея на руках результаты blastn, который в нашем случае демонстрирует, что геномы вообще говоря очень похожи, мы при помощи megablast'а можем понять, какие из участков этих геномов имеют наиболее высокую идентичность, что может, например, говорить об их консервативности и особой функциональной значимости.
Напоследок, посмотрим гифку, повествующую об изменениях `dotplot`'а в зависимости от `word_size`: