Практикум №3
Задание №1. Предсказание вторичной структуры тРНК.
Упражнение 1.
Последовательность тРНК c ID: "1ffy" была получена из fasta файла, скачанного с PDB.
Для предсказания вторичной структуры тРНК была использована программа einverted из пакета EMBOSS. Я использовала программу с такими параметрами:
einverted -sequence rna.seq -gap 12 -threshold 18 -match 5 -mismatch -6 -outfile outfile -outseq seqout
Программа выдала следующее:
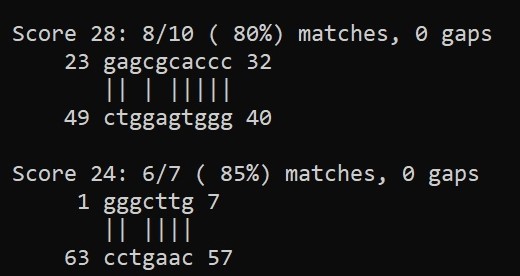
Упражнение 2.
Далее для предсказания вторичной структуры был использован алгоритм Зукера.
На мой взгляд, структура получилась не совсем правдоподобной.
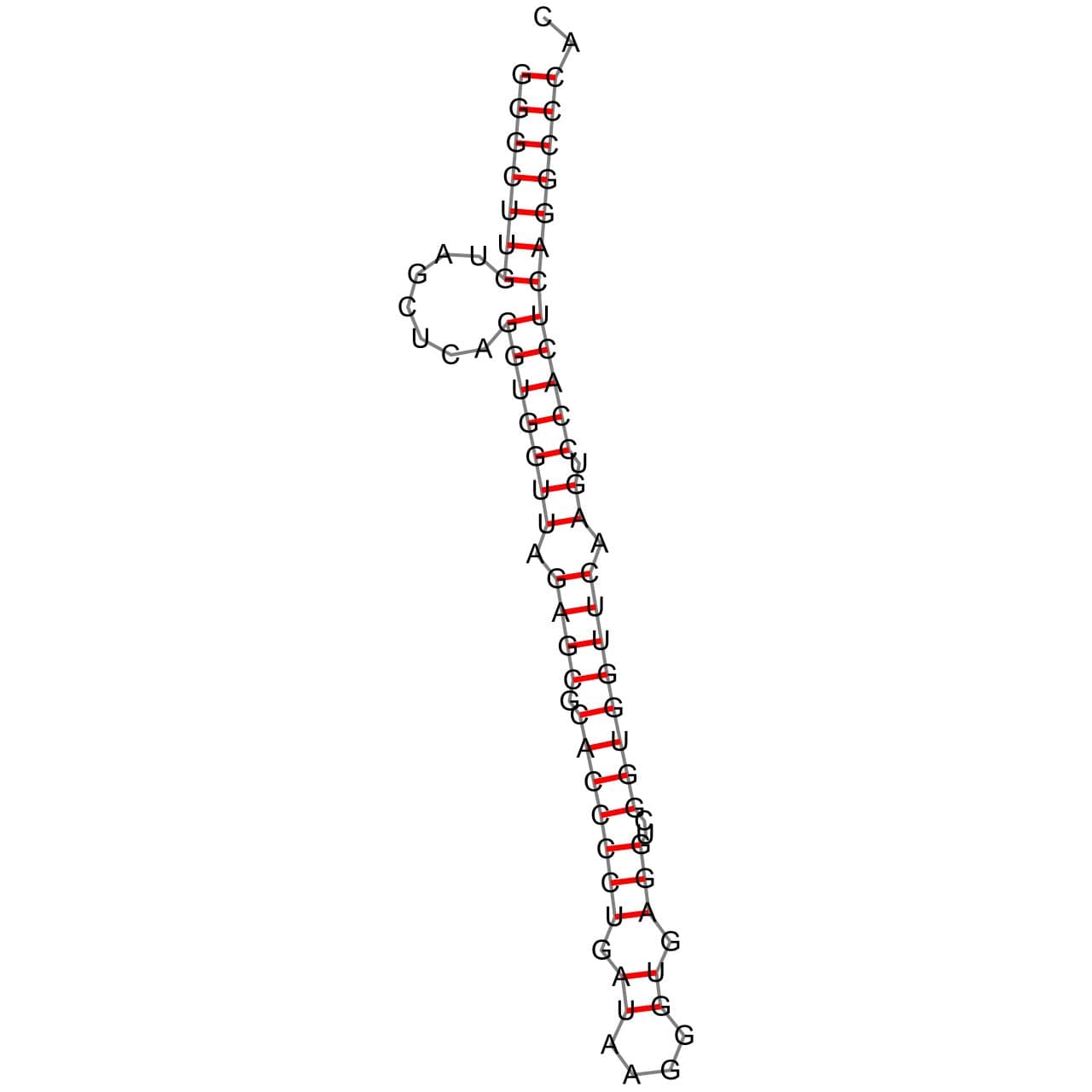
| Участок структуры | Позиции в структуре (по результатам find_pair ) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
|---|---|---|---|
| Акцепторный стебель | 5' 1-7 3' 5' 66-72 3' |
5' 1-7 3' 5' 57-63 3' |
5' 1-7 3' 5' 67-73 3' |
| D-стебель | 5' 10-13 3' 5' 22-25 3' |
- | - |
| T-стебель | 5' 49-53 3' 5' 61-65 3' |
- | - |
| Антикодоновый стебель | 5' 26-32 3' 5' 38-44 3' |
5' 23-32 3' 5' 40-49 3' |
- |
| Общее число канонических пар нуклеотидов | 20 | 14 | 27 |
Результаты предсказания с помощью einverted оказались не самыми удачными. Как бы я ни меняла параметры, находилось лишь 2 стебля. Алгоритм Зукера предсказал неверные стебли в тРНК. Во-первых, их оказалось больше, чем 4. Во-вторых, они совершенно не сопоставлялись тем, что я нашла в find_pairs.
Задание №2. Поиск ДНК-белковых контактов в заданной структуре.
Упражнение 1.
В этом упражнении надо было с помощью команды define JMol:
Упражнение 2.
В этом упражнении нужно было искать контакты ДНК-белок. Для их поиска я создала скрипт-файл. Результаты заношу в таблицу.
Полярным назывался такой контакт, который осуществлялся между полярными атомами (O, N) белка и ДНК на расстоянии меньше 3.5 ангстрем.
Неполярным назывался такой контакт, который осуществлялся между неполярными атомами (С, P, S) белка и ДНК на расстоянии меньше 4.5 ангстрем.
| Контакты атомов белка с | Полярные | Неполярные | Всего |
|---|---|---|---|
| остатками 2'-дезоксирибозы | 1 | 20 | 21 |
| остатками фосфорной кислоты | 13 | 60 | 73 |
| остатками азотистых оснований со стороны большой бороздки | 4 | 9 | 13 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 1 | 1 |
Упражнение 3.
В этом упражнении нужно было получить схему ДНК-белковых контактов с помощью программы nucplot
Т.к программа nucplot работает только со старым форматом файлов PDB, то я перевела скачанный файл в старый формат. Затем запустила программу:
remediator --old ''1r4o.pdb'' > ''1r4o_old.pdb''
nucplot 1r4o_old.pdb
Далее файл nucplot.ps я перевела в формат pdf (с помощью конвертера)
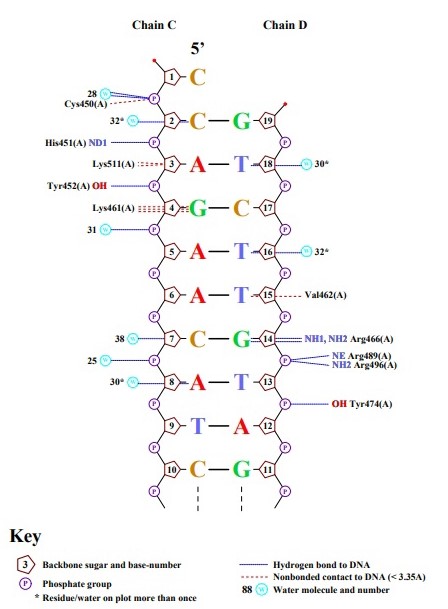
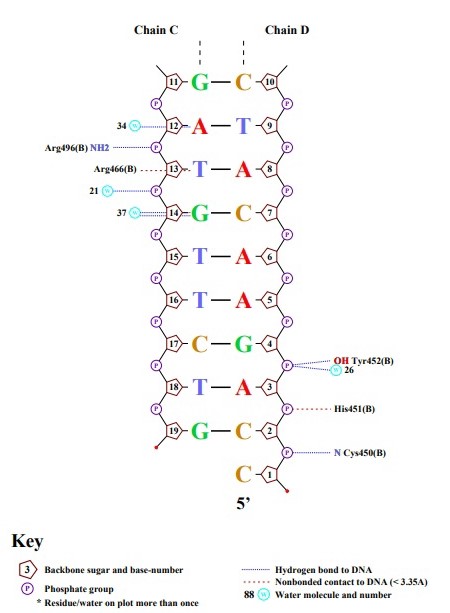
Упражнение 4.
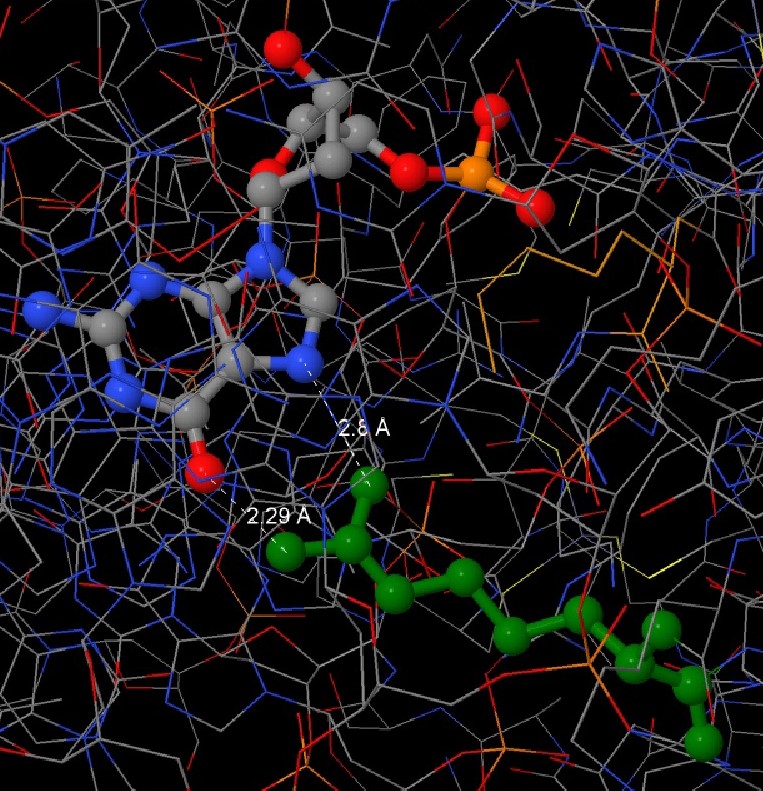
На рисунках выше я выбрала аминокислотный остаток с наибольшим числом указанных на схеме контактов с ДНК - Lys461:A. Он имеет 3 контанка (все они < 3.5 ангстрем) с DG4:C (Рис.2.)
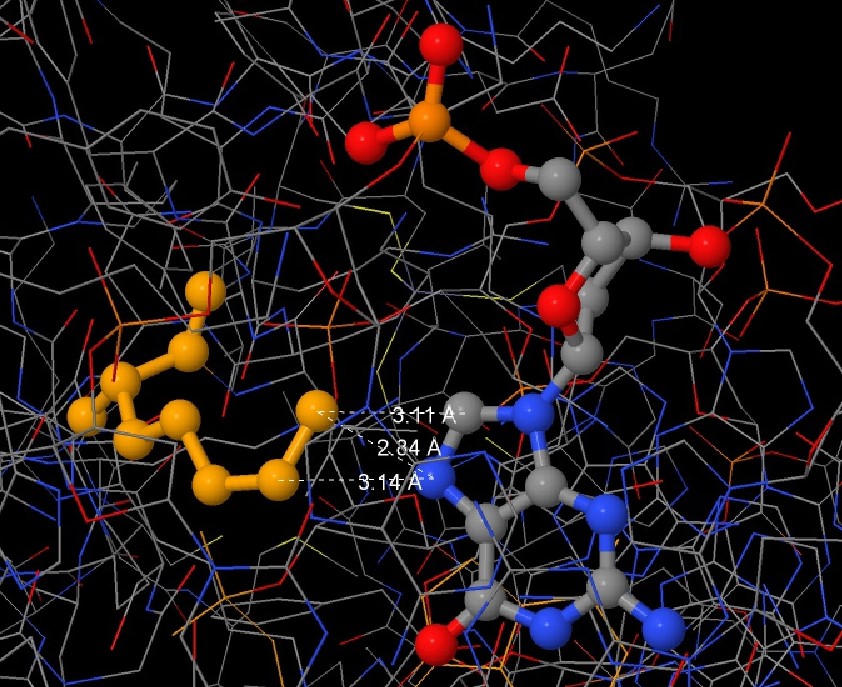
Контакт Arg466:A с DG14:D мне показался важным, т.к возникают 2 водородные связи. (Рис.3.)