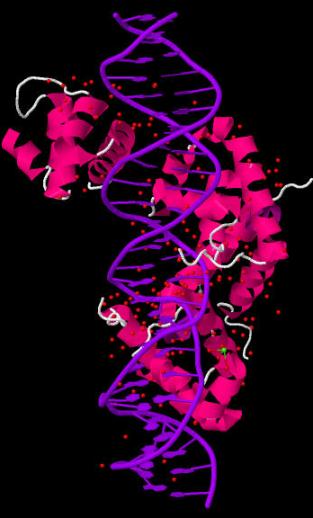
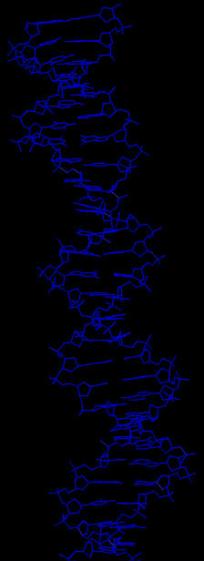
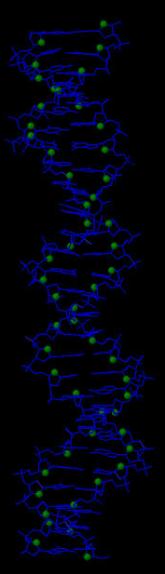
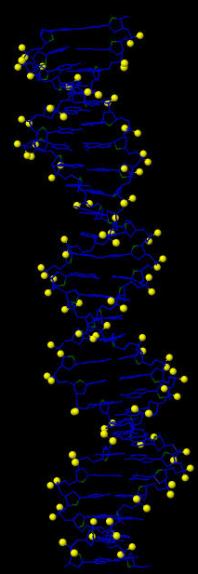
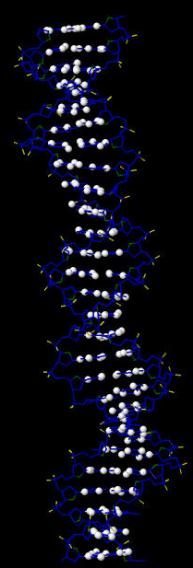
В программе RasMol с помощью команды define определяем и задаем множества следующих атомов:
множество атомов кислорода 2'-дезоксирибозы (set1); множество атомов кислорода в остатке фосфорной кислоты (set2); множество атомов азота в азотистых основаниях(set3);
Напишем скрипт1 с определениями этих множеств.
Далее создаем скрипт2, который даёт в программе RasMol последовательно следующие изображения:
вся структура, только ДНК (в проволочной модели), проволочная модель ДНК с выделенными шариками множества атомов set1, проволочная модель ДНК с выделенными шариками множества атомов set2, проволочная модель ДНК с выделенными шариками множества атомов set3.
Ниже представлены последовательные изображения, полученные с помощью скрипта2:
Будем считать полярными атомы кислорода и азота, а неполярными атомы углерода, фосфора и серы. Назовем полярным контактом ситуацию, в которой расстояние между полярным атомом белка и полярным атомом ДНК меньше 3.5A. Аналогично, неполярным контактом будем считать пару неполярных атомов на расстоянии меньше 4.5A.
Для удобства был составлен скрипт3 последовательно выводящий на экран изображения структуры и атомов, необходимых для определения ДНК-белкового контакта соответствующего типа. Результаты определения числа контактов приведены в таблице ниже:| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 0 | 21 | 21 |
| остатками фосфорной кислоты | 29 | 28 | 57 |
| остатками азотистых оснований со стороны большой бороздки | 17 | 34 | 51 |
| остатками азотистых оснований со стороны малой бороздки | 0 | 3 | 3 |
Видно, что число неполярных взаимодействий намного превышает число полярных, вероятно, это связано с тем, что полярных атомов меньше, и большинство из них скрыто внутри молекул. Не обнаружено полярных контактов между белком и остатками азотистых оснований со стороны малой бороздки, скорее всего это вызвано пространственной недоступностью атомов азотистых оснований, направленных в сторону малой бороздки.
Для получения популярной схемы ДНК-белковых контактов использовали программу nucplot. Эта программа работает только со старым форматом .pdb, поэтому для работы с ней был использован файл 1rio_old.pdb, полученный с помощью программы remediator:
Одним из полученных файлов является файл nucplot.ps, содержание которого представлено на изображении ниже:
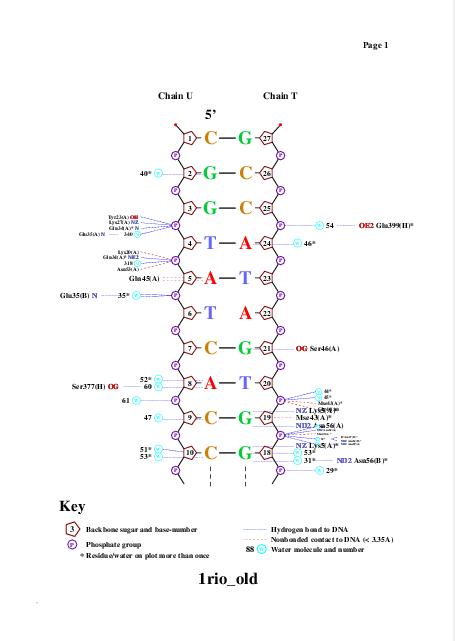
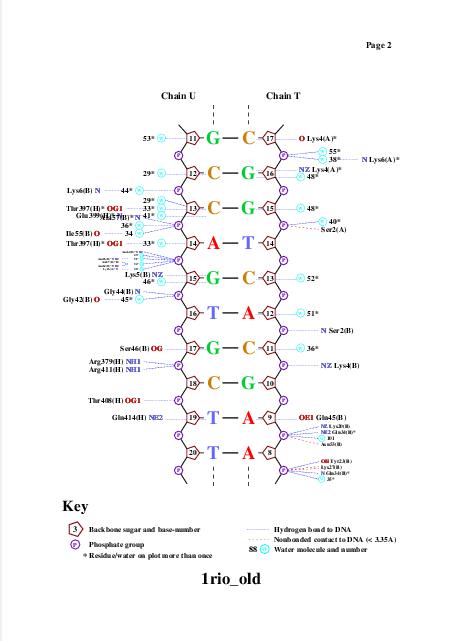
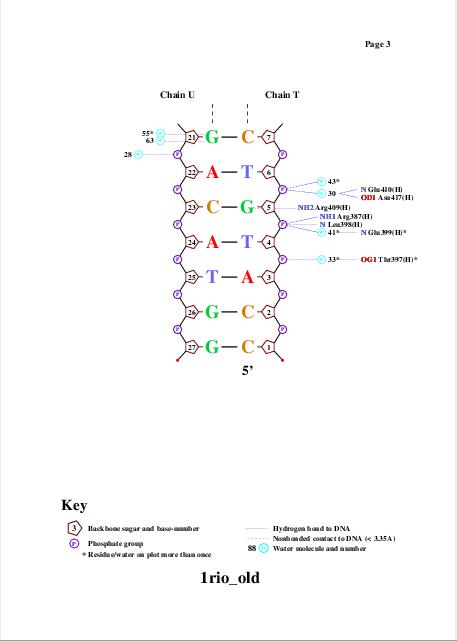
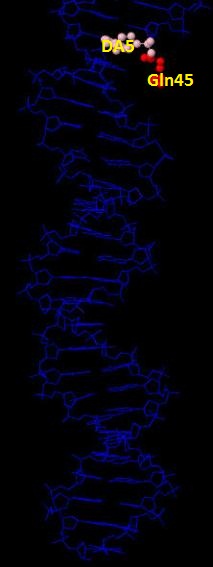
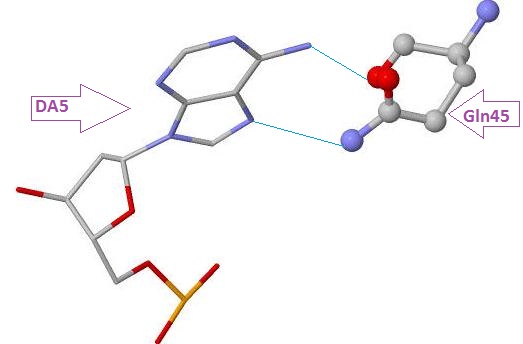
Glu399 и Thr397 - аминокислотные остатки с наибольшим числом указанных на схеме контактов - 3. Но они координируют молекулы воды, а не напрямую взаимодействуют с атомами ДНК. Для участия в распознавании последовательности ДНК аминокислотный остаток, очевидно, должен связываться с азотистым основанием. Как мы видим на схеме ДНК-белковых контактов, таких остатков не так много, я остановлюсь всего на двух: Gln45 и Arg409, первый из них образует 2 водородные связи с атомами азотистого основания DA5, поэтому я предположу, что именно Gln45 является наиболее важным в распознавании последовательности ДНК. Ниже приведены иллюстрации взаимодействия Gln45 c DA5.
Программа einverted из пакета EMBOSS позволяет найти инвертированные участки в нуклеотидных последовательностях. Найдём возможные комплементарные участки в последовательности исследуемой тРНК (последовательность тРНК в fasta-формате можно найти в файле 1F7V.fasta) используя команду:
einverted 1F7V.fasta
При параметрах по умолчанию инвертированных участков найдено не было. Пришлось подобирать наиболее удачные параметры :
Gap penalty: 12 Minimum score threshold: 15 Match score: 3 Mismatch score: 0 Для антикодонового и D стеблей
Gap penalty: 0 Minimum score threshold: 10 Match score: 2 Mismatch score: -2 Для акцепторного стебля
Алгоритм Зукера выполняет программа mfold. Мы изменяем только параметр P, указывающий, на сколько процентов выдаваемое предсказание структуры может отличаться по своей вычисленной энергии от оптимального. Пришлось использовать программу mfold онлайн на сервере Mobyle @Pasteur.
Применение программы mfold для последовательности 1F7V.fasta с минимальным параметром P = 15 приводит к получению удачного предсказания, близкого к реальной структуре. Мы можем наблюдать предсказание вторичной структуры РНК по алгоритму Зукера на рисунке ниже.
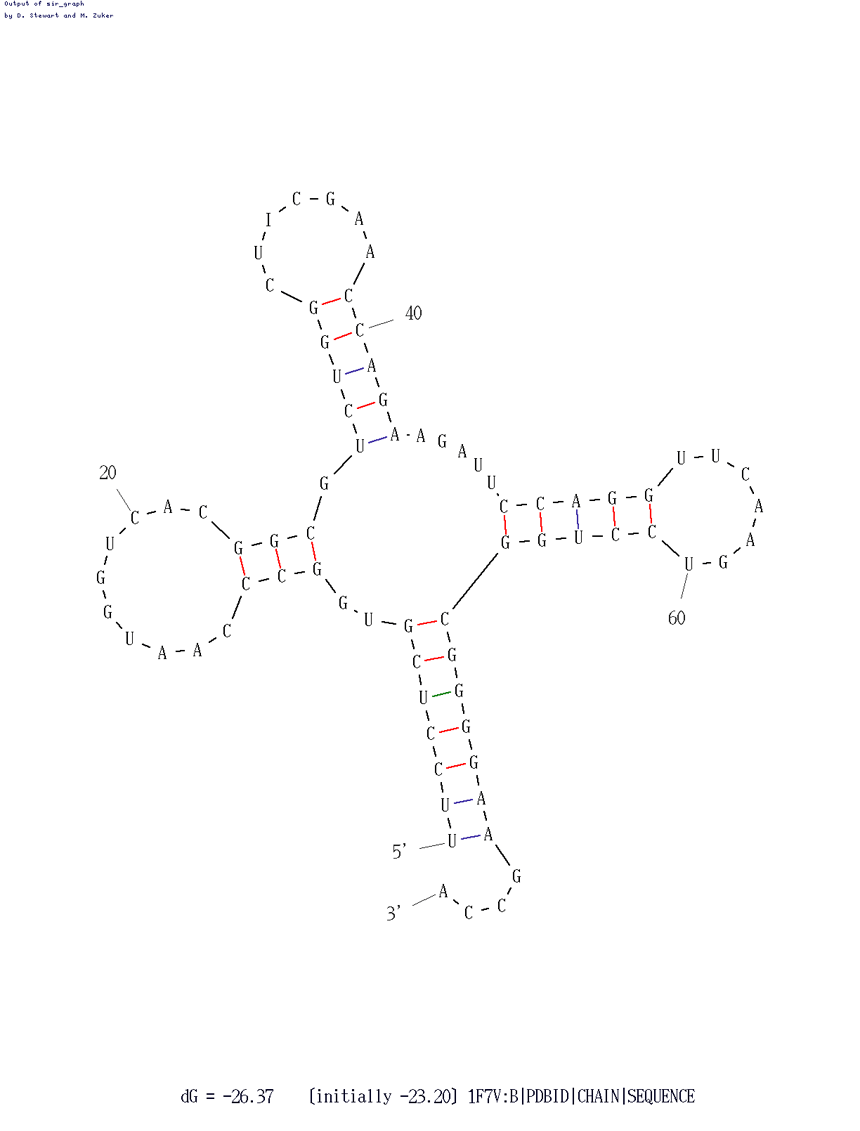
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted | Результаты предсказания по алгоритму Зукера |
| Акцепторный стебель | 5'-901-907-3' 5'-966-972-3' Всего 7 пар | 2 пары из 7 предсказаны | 7 пар из 7 реальных |
| T-стебель | 5'-949-953-3' 5'-961-965-3' Всего 5 пар | 5 пар из 5 реальных | 5 пар из 5 реальных |
| D-стебель | 5'-910-913-3' 5'-922-925-3' Всего 4 пар | Нет предсказаний | 3 пары из 4 реальных |
| Антикодоновый стебель | 5'-939-944-3' 5'-926-931-3' Всего 6 пар | 5 пар из 6 реальных | 5 пар из 6 реальных |
| Общее число канонических пар нуклеотидов | 13 | 12 | 19 |
Наиболее точными способами определения вторичной структуры тРНК, на мой взгляд, являются программа find_pair и алгоритм Зукера. При помощи einverted можно предсказать только часть вторичной структуры тРНК, а именно программа рассматривает исключительно канонические взаимодействия, причём результаты сильно меняются в зависимости от введения разных параметров.