Ресеквенирование
Практикум был выполнен для восьмой хромосомы.
| копирование файла | cp ../Human/chr8.fasta . |
|---|---|
| индексирование файла | hisat2-build chr8.fasta index |
| копирование файла [2] | cp ../Human/reads/chr8.fastq . |
| анализ качества чтений | fastqc chr8.fastq |
| очистка чтений | java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr8.fastq chr8_trimmed.fastq TRAILING:20 MINLEN:50 |
| анализ качества чтений [2] | fastqc chr8_trimmed.fastq |
| картирование чтений | hisat2 -x index -U chr8_trimmed.fastq -S chr8.sam --no-softclip --no-spliced-alignment |
| перевод в .bam | samtools view -b -o chr8.bam chr8.sam |
| сортировка чтений | samtools sort chr8.bam chr8_sorted |
| индексирование файла [2] | samtools index chr8_sorted.bam |
| создание файла с полиморфизмами | samtools mpileup -uf chr8.fasta -o polymorph.bcf chr8_sorted.bam |
| изменение формата | bcftools call -cv -o polymorph.vcf polymorph.bcf |
| удаление инделей | vcftools --vcf polymorph.vcf --remove-indels --recode --out snp |
| изменение формата [2] | convert2annovar.pl -format vcf4 snp.recode.vcf -outfile snp.avinput |
| аннотация (refgene) | annotate_variation.pl -out snp_refgene -build hg19 -dbtype refGene snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| аннотация (dbsnp) | annotate_variation.pl -filter -out snp_dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| аннотация (1000 genomes) | annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out snp_1000genomes snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| аннотация (GWAS) | annotate_variation.pl -regionanno -build hg19 -out snp_gwas -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
| аннотация (clinvar) | annotate_variation.pl -filter -dbtype clinvar_20150629 -buildver hg19 -out snp_clinvar snp.avinput /nfs/srv/databases/annovar/humandb.old/ |
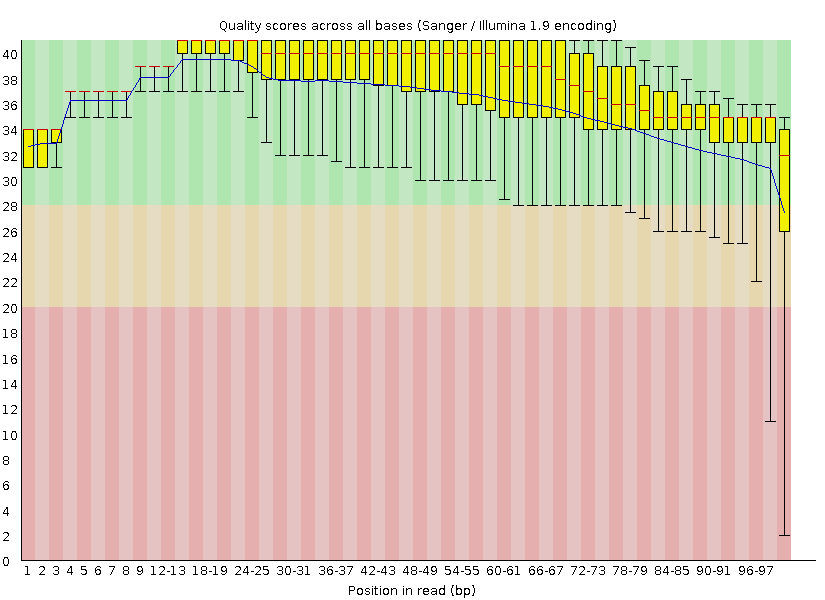
Результат работы команды Trimmomatic:
Input Reads: 8367 Surviving: 8227 (98,33%) Dropped: 140 (1,67%)
То есть изначально было 8367 чтений.
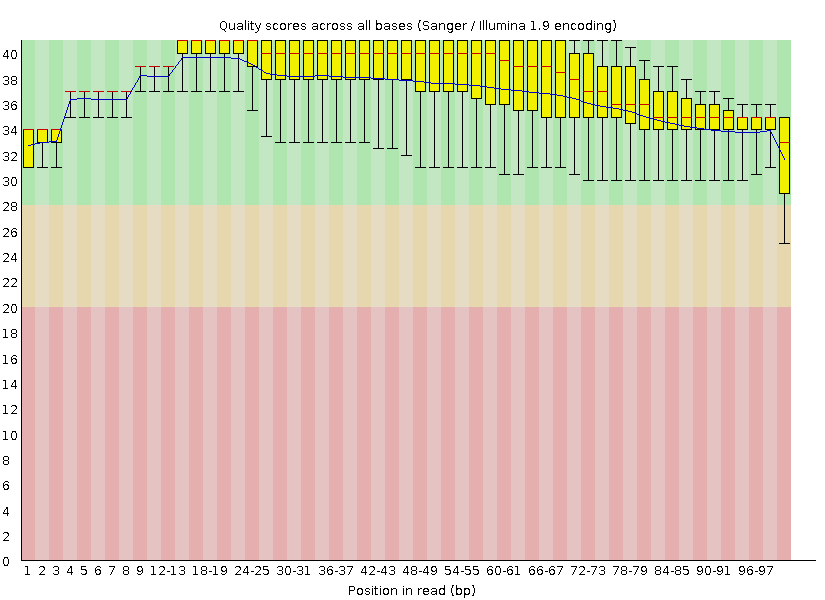
Скорее всего можно было обойтись и без триммирования. У чтений изначально высокое качество, и только усы двух последних боксплотов заползают в красную зону. Чуть больше полутора процентов чтений было удалено - такое количество (учитывая изначально неплохое качество) вряд ли могло сильно изменить последующий анализ.
Результат работы команды hisat2:
8227 reads; of these:
8227 (100.00%) were unpaired; of these:
30 (0.36%) aligned 0 times
8197 (99.64%) aligned exactly 1 time
0 (0.00%) aligned >1 times
99.64% overall alignment rate
На геном откартировалось 99,64% чтений (причем каждое - не более одного раза). Это очень хорошее качество.
| строчка в файле polymorph.vcf | 33 | 42 | 90 |
|---|---|---|---|
| координата | 27454785 | 27467821 | 116424270 |
| тип | делеция | замена | вставка |
| референс | TAATGAA | C | A |
| чтение | TAA | G | AC |
| глубина покрытия | 5 | 13 | 28 |
| качество чтения | 58.4663 | 149.008 | 57.456 |
Всего получается 95 SNP и 5 инделей.
| качество | глубина | |
|---|---|---|
| среднее | 66.05637 | 14.87 |
| первый квантиль | 7.79993 | 1 |
| медиана | 11.3429 | 2 |
| третий квантиль | 113.37025 | 16.25 |
Всего в RefSeq девять категорий SNP, но в этом случае представлены только четыре:
| exonic | 5 |
|---|---|
| intergenic | 17 |
| intronic | 60 |
| UTR3 | 13 |
Отсутствуют splicing, ncRNA, UTR5, upstream и downstream.
| ген | референс | чтение | тип замены |
|---|---|---|---|
| CLU | A (H) | G (H) | синонимичная |
| HNF4G | G (S) | A (N) | несинонимичная |
| HNF4G | G (L) | A (L) | синонимичная |
| HNF4G | G (M) | A (I) | несинонимичная |
| TRPS1 | C (P) | A (P) | синонимичная |
То есть SNP попали в три гена. Если посмотреть выдачу по dnsnp, rs имеют 77 SNP из 95. Средняя частота SNP по 1000genomes равна 0.4633. Первый, второй и третий квантили - 0.0718, 0.5382 и 0.6744 соответственно. При этом для экзонных SNP средняя частота составляет 0.4129. Выглядит достаточно безобидно, если не посмотреть, что у несинонимичных замен частоты равны 0.6118 и 0.6148. Здравый смысл подсказывает, что все должно быть наоборот. Скорее всего просто маленькая выборка.
Что касается клинической аннотации, она имеется у четырех SNP. Первые два - в интронах гена CLU, кодирующего кластерин. Одна из его функций - регуляция апоптоза. Его оверэкспрессия защищает клетки в случае стресса, поэтому ассоциированность с болезнью Альцгеймера - нейродегенеративным заболеванием! - выглядит довольно логично. Мутация в UTR3 гена, который кодирует ядерный фактор гепатоцитов 4 гамма, менее очевидна. Он, кажется, связывает жирные кислоты, и непонятно, как он может повлиять на уровень уратов. С геном TRPS1 (кодирует фактор транскрипции цинкового пальца) все столь же туманно: сложно сказать, какое отношение он может иметь к уровню "хорошего" холестерина. Интересно, что аннотация имеется у тех же трех генов, в которых были найдены экзонные SNP. Совпадение?
P.S. Выдача Clinvar не особо понятная. Зачем она вообще была нужна? Клиническую аннотацию можно (и нужно) смотреть по GWAS.