Практикум 12. Алгоритмы и программы множественного выравнивания
В данной работе были выполнены два задания. В первом задании была реализована программа для сравнения двух множественных выравниваний. Во втором задании эта программа была применена для сравнения выравниваний одних и тех же белковых последовательностей, полученных тремя разными программами MSA.
Задание 1. Реализация программы для сравнения двух множественных выравниваний
Ссылка на программу
Программа для сравнения выравниваний: compare_alignments.py.
Цель задания 1
Целью первого задания была реализация программы, которая сравнивает два множественных выравнивания одних и тех же последовательностей и определяет, какие колонки в этих выравниваниях являются одинаково выровненными.
Описание входных данных
Программа принимает на вход два файла с множественными выравниваниями в формате FASTA. Каждый файл должен содержать один и тот же набор последовательностей. Последовательности внутри одного файла должны быть уже выровнены, то есть иметь одинаковую длину с учётом гэпов.
Названия последовательностей в двух сравниваемых файлах должны совпадать. Порядок последовательностей в файлах не влияет на результат, так как программа дополнительно сортирует названия последовательностей перед сравнением.
Описание алгоритма
Сначала программа считывает оба FASTA-файла и сохраняет данные в словари, где ключом является название последовательности, а значением — соответствующая выровненная последовательность.
Затем выполняется проверка корректности входных данных. Программа проверяет, что в каждом файле все последовательности имеют одинаковую длину. Также проверяется, что в двух файлах содержится один и тот же набор последовательностей. Это необходимо, так как сравнение разных наборов последовательностей было бы некорректным.
Для корректного сравнения программа использует не простое посимвольное сравнение колонок, а сравнение положения остатков в исходных последовательностях. Для каждой колонки строится специальная сигнатура. В этой сигнатуре для каждой последовательности записывается номер аминокислотного остатка в исходной последовательности. Если в данной последовательности в рассматриваемой колонке находится гэп, в сигнатуру записывается символ гэпа.
Две колонки из разных выравниваний считаются одинаково выровненными, если их сигнатуры совпадают. Такой подход позволяет сравнивать именно взаимное расположение аминокислотных остатков, а не только символы в конкретных позициях файлов.
После нахождения всех одинаково выровненных колонок программа выделяет блоки совпадений. Блоком считается участок, где одинаково выровненные колонки идут подряд в обоих выравниваниях. Например, если совпадают пары колонок (5, 7), (6, 8), (7, 9), то это один блок длиной 3. В данной работе минимальная длина блока была принята равной 2.
Запуск программы и формат результата
Программа запускается из командной строки. Пример запуска:
python3 compare_alignments.py A_clustal.fasta B_mafft.fasta -o A_vs_B.tsv
Для сравнения другой пары файлов используется аналогичная команда:
python3 compare_alignments.py A_clustal.fasta C_muscle.fasta -o A_vs_C.tsv
Программа выводит на экран основную статистику сравнения и сохраняет подробные результаты в TSV-файл. В результат входят:
- названия сравниваемых файлов;
- число последовательностей;
- длина первого выравнивания;
- длина второго выравнивания;
- число одинаково выровненных колонок;
- процент одинаково выровненных колонок от длины первого выравнивания;
- процент одинаково выровненных колонок от длины второго выравнивания;
- список одинаково выровненных колонок;
- список блоков одинаково выровненных колонок;
- список одинаковых колонок, не входящих в блоки.
Формат TSV удобен для дальнейшей проверки и анализа, так как такой файл можно открыть в текстовом редакторе, Excel, Google Sheets или другой табличной программе.
Вывод по заданию 1
В результате была реализована программа для сравнения двух множественных выравниваний. Программа запускается из командной строки, проверяет корректность входных данных, сравнивает положения остатков в исходных последовательностях, находит одинаково выровненные колонки, выделяет блоки совпадений и сохраняет результаты в формате TSV. Таким образом, программа соответствует требованиям первого задания.
Задание 2. Сравнение множественных выравниваний, полученных разными программами
Цель задания 2
Целью второго задания было сравнить множественные выравнивания одних и тех же аминокислотных последовательностей, полученные с помощью трёх разных программ, и оценить, насколько похожи результаты этих программ между собой.
Исходные данные
Для анализа были выбраны шесть белковых последовательностей:
- NCK1_HUMAN/1-377;
- VAV_HUMAN/1-845;
- BLK_MOUSE/1-499;
- FGR_HUMAN/1-529;
- LCK_CHICK/1-508;
- HCK_HUMAN/1-526.
Для этих последовательностей были построены три множественных выравнивания:
- A — выравнивание, полученное программой Clustal Omega;
- B — выравнивание, полученное программой MAFFT;
- C — выравнивание, полученное программой MUSCLE.
Сравнение выполнялось для пар A–B и A–C, так как по условию задания нужно было определить, какое из выравниваний, B или C, больше похоже на выравнивание A.
Ссылки на файлы
- A_clustal.fasta — выравнивание Clustal Omega;
- B_mafft.fasta — выравнивание MAFFT;
- C_muscle.fasta — выравнивание MUSCLE;
- A_vs_B.tsv — результат сравнения A и B;
- A_vs_C.tsv — результат сравнения A и C;
- проект Jalview со сравниваемыми выравниваниями.
Проверка входных данных
Перед сравнением было проверено, что все три выравнивания содержат один и тот же набор последовательностей. Во всех трёх файлах присутствуют шесть последовательностей: NCK1_HUMAN/1-377, VAV_HUMAN/1-845, BLK_MOUSE/1-499, FGR_HUMAN/1-529, LCK_CHICK/1-508 и HCK_HUMAN/1-526.
Длины последовательностей без гэпов во всех трёх выравниваниях совпадают и составляют 377, 845, 499, 529, 508 и 526 аминокислот соответственно. Это подтверждает, что сравнивались разные выравнивания одного и того же набора белковых последовательностей.
Метод сравнения
Сравнение выравниваний было выполнено с помощью программы, написанной в первом задании. Для каждой колонки каждого выравнивания программа определяла, какие именно аминокислотные остатки исходных последовательностей находятся в данной колонке. Две колонки из разных выравниваний считались одинаково выровненными, если в них совпадали положения остатков во всех последовательностях.
Кроме общего списка одинаково выровненных колонок, программа выделяла блоки совпадений. Блоком считался непрерывный участок, в котором одинаково выровненные колонки шли подряд в обоих сравниваемых выравниваниях. Минимальная длина блока составляла 2 колонки.
Команды для запуска сравнения
python3 compare_alignments.py A_clustal.fasta B_mafft.fasta -o A_vs_B.tsv python3 compare_alignments.py A_clustal.fasta C_muscle.fasta -o A_vs_C.tsv
Результаты сравнения A и B
В первой паре сравнивались выравнивания A, полученное программой Clustal Omega, и B, полученное программой MAFFT.
| Параметр | Значение |
|---|---|
| Файл 1 | A_clustal.fasta |
| Файл 2 | B_mafft.fasta |
| Число последовательностей | 6 |
| Длина выравнивания A | 1141 колонка |
| Длина выравнивания B | 1002 колонки |
| Число одинаково выровненных колонок | 161 |
| Доля одинаковых колонок от длины A | 14.11% |
| Доля одинаковых колонок от длины B | 16.07% |
Для пары A–B были найдены следующие блоки одинаково выровненных колонок:
(58,110)-(61,113), length=4;
(99,182)-(112,195), length=14;
(129,217)-(145,233), length=17;
(194,293)-(210,309), length=17;
(221,320)-(228,327), length=8;
(293,421)-(320,448), length=28;
(340,468)-(341,469), length=2;
(352,480)-(363,491), length=12;
(393,522)-(405,534), length=13;
(410,539)-(412,541), length=3;
(422,551)-(431,560), length=10;
(466,597)-(472,603), length=7;
(506,648)-(513,655), length=8;
(638,770)-(639,771), length=2;
(689,820)-(690,821), length=2;
(806,929)-(813,936), length=8.
Одинаково выровненные колонки, не входящие в блоки:
Результаты сравнения A и C
Во второй паре сравнивались выравнивания A, полученное программой Clustal Omega, и C, полученное программой MUSCLE.
| Параметр | Значение |
|---|---|
| Файл 1 | A_clustal.fasta |
| Файл 2 | C_muscle.fasta |
| Число последовательностей | 6 |
| Длина выравнивания A | 1141 колонка |
| Длина выравнивания C | 893 колонки |
| Число одинаково выровненных колонок | 175 |
| Доля одинаковых колонок от длины A | 15.34% |
| Доля одинаковых колонок от длины C | 19.60% |
Для пары A–C были найдены следующие блоки одинаково выровненных колонок:
(106,136)-(109,139), length=4;
(117,147)-(120,150), length=4;
(143,175)-(159,191), length=17;
(181,213)-(183,215), length=3;
(200,232)-(204,236), length=5;
(232,268)-(234,270), length=3;
(262,298)-(283,319), length=22;
(285,321)-(305,341), length=21;
(321,357)-(349,385), length=29;
(359,395)-(379,415), length=21;
(390,426)-(393,429), length=4;
(462,504)-(472,514), length=11;
(476,518)-(484,526), length=9;
(507,549)-(512,554), length=6;
(519,561)-(522,564), length=4;
(638,655)-(639,656), length=2;
(811,825)-(813,827), length=3;
(928,498)-(929,499), length=2;
(1040,718)-(1041,719), length=2.
Одинаково выровненные колонки, не входящие в блоки:
Примеры совпадающих и различающихся участков
При сравнении A и B можно выделить несколько хорошо выраженных совпадающих участков: (99,182)-(112,195), (129,217)-(145,233) и (293,421)-(320,448). Эти блоки показывают, что в данных областях Clustal Omega и MAFFT расположили остатки последовательностей одинаковым образом.
Для пары A и C также были найдены протяжённые совпадающие участки: (262,298)-(283,319), (285,321)-(305,341), (321,357)-(349,385) и (359,395)-(379,415). Особенно заметен блок длиной 29 колонок: (321,357)-(349,385).
Различающиеся участки выявляются по разрывам между блоками совпадений и по наличию одиночных совпадающих колонок, которые не входят в блоки. Например, в паре A–B одиночными совпадающими колонками являются (374,502) и (998,665), а в паре A–C — (405,441). Это означает, что в соседних областях программы по-разному расставили гэпы и получили различное взаимное расположение остатков.
Таким образом, по результатам программного сравнения можно указать не менее трёх участков совпадения и не менее двух участков несовпадения, что соответствует требованиям задания. Совпадающие участки представлены протяжёнными блоками одинаково выровненных колонок, а несовпадающие участки — промежутками между такими блоками и одиночными совпадающими колонками.
Обсуждение результатов
Полученные выравнивания заметно различаются по длине. Самым длинным оказалось выравнивание A, построенное программой Clustal Omega: оно содержит 1141 колонку. Выравнивание B, полученное программой MAFFT, содержит 1002 колонки, а выравнивание C, полученное программой MUSCLE, содержит 893 колонки.
Различие в длине выравниваний связано с тем, что программы используют разные алгоритмы построения множественного выравнивания и по-разному вставляют гэпы. Особенно сильно это проявляется при анализе белков разной длины: в данном наборе самая короткая последовательность содержит 377 аминокислот, а самая длинная — 845 аминокислот.
При сравнении A и B было найдено 161 одинаково выровненное положение, что составляет 14.11% от длины выравнивания A и 16.07% от длины выравнивания B. При сравнении A и C было найдено 175 одинаково выровненных положений, что составляет 15.34% от длины выравнивания A и 19.60% от длины выравнивания C.
Следовательно, по общему числу одинаково выровненных колонок выравнивание C оказалось немного ближе к A, чем выравнивание B. Кроме того, в паре A–C наблюдаются несколько длинных блоков совпадений, включая блоки длиной 22, 21, 29 и 21 колонка. В паре A–B самый длинный блок имеет длину 28 колонок, однако общее число совпадающих колонок меньше.
Тем не менее процент совпадающих колонок в обоих сравнениях остаётся относительно невысоким. Это показывает, что результат множественного выравнивания существенно зависит от выбранной программы. Вероятно, основные различия связаны с вариабельными участками белков, вставками, делециями и различиями в длине анализируемых последовательностей.
Вывод по заданию 2
В ходе второго задания были построены и сравнены три множественных выравнивания одного и того же набора из шести белковых последовательностей. Выравнивание A было получено программой Clustal Omega, выравнивание B — программой MAFFT, а выравнивание C — программой MUSCLE.
Сравнение было выполнено для пар A–B и A–C с помощью программы, реализованной в первом задании. Для пары A–B было найдено 161 одинаково выровненное положение, а для пары A–C — 175 одинаково выровненных положений. Таким образом, по использованному критерию выравнивание MUSCLE оказалось немного более похожим на выравнивание Clustal Omega, чем выравнивание MAFFT.
Полученные результаты соответствуют требованиям задания: были использованы три разные программы MSA, выполнено сравнение A–B и A–C, получены таблицы результатов в формате TSV, выделены блоки одинаково выровненных колонок, указаны одинаковые колонки, не входящие в блоки, приведены примеры совпадающих и различающихся участков, а также сделан вывод о том, какое из выравниваний больше похоже на A.
Задание 3. Построение выравнивания по совмещению структур и его сравнение с ClustalO
Цель задания 3
Целью третьего задания было построить выравнивание аминокислотных последовательностей на основе пространственного совмещения белковых структур и сравнить его с множественным выравниванием тех же последовательностей, полученным с помощью программы множественного выравнивания.
В отличие от обычного множественного выравнивания последовательностей, структурное выравнивание основано не только на сходстве аминокислотных остатков, но и на взаимном расположении атомов в пространстве. Поэтому такое выравнивание позволяет оценить, насколько последовательностное выравнивание согласуется с пространственной организацией белков.
Исходные данные
Для анализа были выбраны три белковые структуры, содержащие сходные доменные участки:
- 2CUB;
- 4HCK;
- 1KIK.
Для этих структур были подготовлены два выравнивания:
- structure_alignment.fasta — выравнивание последовательностей, полученное из структурного совмещения;
- ClustalO_pdb_sequences.fasta — множественное выравнивание тех же последовательностей, полученное в Jalview c помощью Clustal Omega.
Также для проверки и визуализации были использованы следующие файлы:
- structure_superposition.png — изображение совмещения трёх структур;
- structure_vs_ClustalO.tsv — результат программного сравнения структурного выравнивания и ClustalO;
- pr12_task3_comparison.jvp — проект Jalview со сравниваемыми выравниваниями;
- 2CUB.pdb, 4HCK.pdb, 1KIK.pdb — PDB-файлы использованных структур.
Получение изображения совмещения структур
Пространственное совмещение структур было выполнено в PyMOL. В качестве референсной структуры была использована структура 2CUB, относительно которой были совмещены структуры 4HCK и 1KIK.
Для визуализации были использованы следующие команды PyMOL:
load 2CUB.pdb load 4HCK.pdb load 1KIK.pdb align 4HCK, 2CUB align 1KIK, 2CUB hide everything show cartoon color green, 2CUB color blue, 4HCK color orange, 1KIK bg_color white orient zoom ray 1600, 1200 png structure_superposition.png, dpi=300
Полученное изображение показывает, что центральные части доменов хорошо совмещаются между собой. Наиболее консервативные элементы вторичной структуры располагаются близко в пространстве, тогда как периферические петлевые и концевые участки отличаются сильнее.
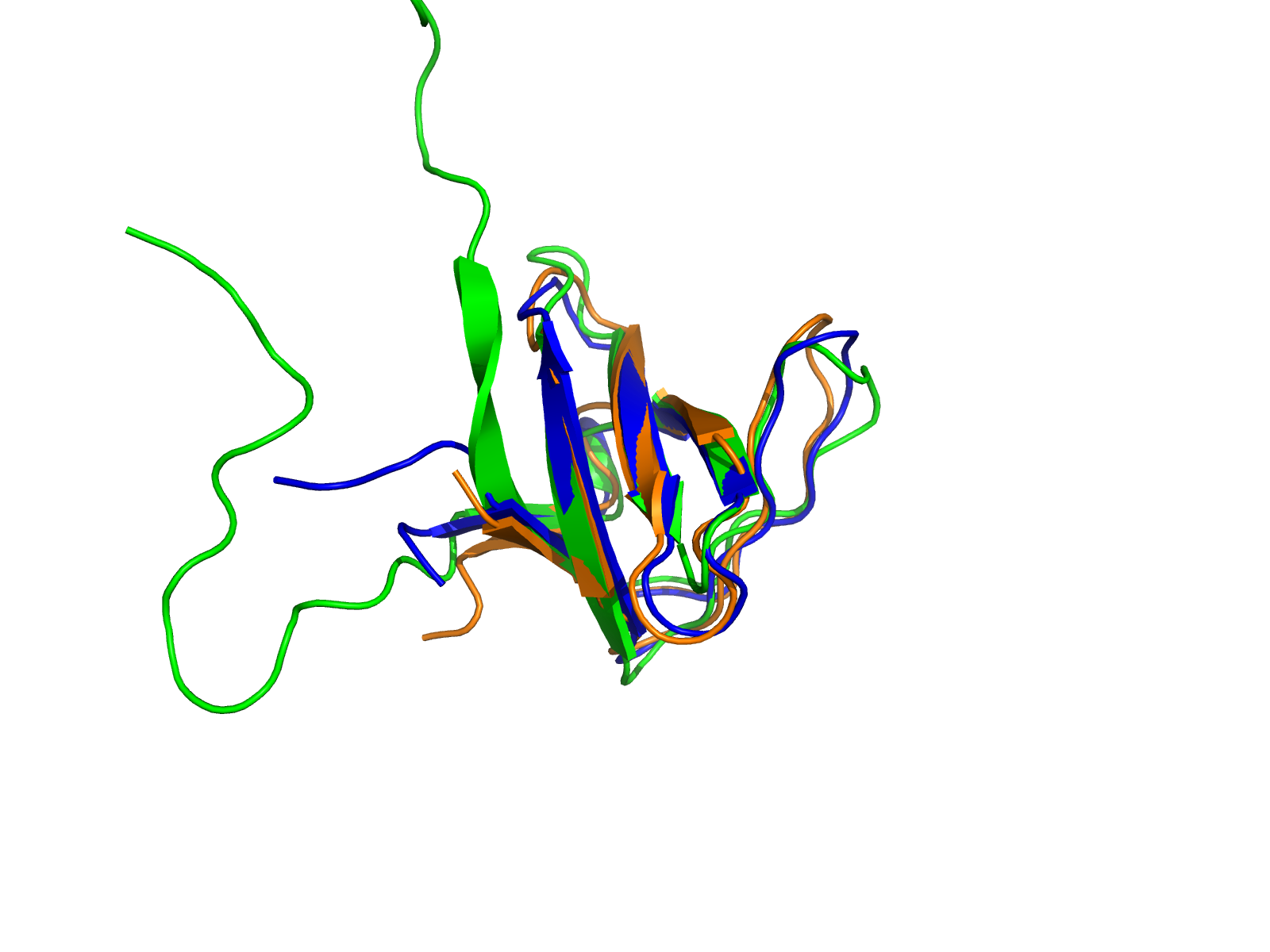
Описание структурного выравнивания
Структурное выравнивание было получено из совмещения трёх белковых структур. В таком выравнивании в одной колонке располагаются аминокислотные остатки, Cα-атомы которых занимают близкие положения после пространственного совмещения структур.
Для сравнения использовались последовательности из PDB-файлов, а не полные записи UniProt. Это важно, так как последовательность белка в PDB-файле может соответствовать только фрагменту полной белковой последовательности. Поэтому для корректного сравнения оба выравнивания должны содержать один и тот же набор остатков.
Проверка выравниваний перед сравнением
Перед программным сравнением оба FASTA-файла были приведены к одинаковому виду: названия последовательностей совпадали, а буквы аминокислотных остатков были приведены к верхнему регистру. Это необходимо, поскольку программа сравнения проверяет не только расположение гэпов, но и совпадение исходных последовательностей без гэпов.
В сравниваемых файлах присутствовали три последовательности: 2CUB, 4HCK и 1KIK. Таким образом, сравнение выполнялось для одного и того же набора последовательностей, что соответствует требованиям задания.
Метод сравнения структурного выравнивания и ClustalO
Сравнение было выполнено с помощью программы compare_alignments.py, реализованной в первом задании. Программа сравнивает не просто символы в колонках, а положение остатков исходных последовательностей. Для каждой колонки строится сигнатура, в которой для каждой последовательности указано, какой аминокислотный остаток находится в данной колонке. Если в колонке стоит гэп, в сигнатуру записывается символ гэпа.
Две колонки из разных выравниваний считаются одинаково выровненными, если их сигнатуры совпадают. Такой подход позволяет корректно сравнивать выравнивания, даже если они имеют разную длину и разное расположение гэпов.
Результаты программного сравнения
В результате сравнения структурного выравнивания и выравнивания ClustalO были получены следующие значения:
| Параметр | Значение |
|---|---|
| Файл 1 | structure_alignment_upper.fasta |
| Файл 2 | ClustalO_pdb_sequences_upper.fasta |
| Число последовательностей | 3 |
| Длина структурного выравнивания | 91 колонка |
| Длина выравнивания ClustalO | 90 колонок |
| Число одинаково выровненных колонок | 86 |
| Доля одинаковых колонок от длины структурного выравнивания | 94.51% |
| Доля одинаковых колонок от длины ClustalO | 95.56% |
| Минимальная длина блока | 2 колонки |
Были найдены следующие блоки одинаково выровненных колонок:
(33,32)-(57,56), length=25;
(61,60)-(91,90), length=31.
Одинаково выровненная колонка, не входящая в блок:
Список одинаково выровненных колонок, не входящих в блоки
В результате сравнения была найдена одна одинаково выровненная колонка, которая не входит в блоки:
| Колонка в структурном выравнивании | Колонка в ClustalO |
|---|---|
| 59 | 58 |
Эта колонка совпадает в двух выравниваниях, однако соседние колонки вокруг неё выровнены по-разному. Поэтому программа не включает её в блок, так как минимальная длина блока была задана равной двум подряд идущим колонкам.
Примеры совпадающих и различающихся участков
По результатам сравнения можно выделить несколько крупных совпадающих участков. Первый блок, (1,1)-(29,29), имеет длину 29 колонок. Второй блок, (33,32)-(57,56), имеет длину 25 колонок. Третий блок, (61,60)-(91,90), имеет длину 31 колонку. Эти блоки показывают, что большая часть структурного выравнивания и выравнивания ClustalO совпадает.
Несовпадающие участки расположены между найденными блоками. Первый участок различий находится между колонками 29 и 33 структурного выравнивания, то есть в области перехода от первого блока совпадений ко второму. Второй участок различий находится между колонками 57 и 61 структурного выравнивания. Именно в этих областях два способа выравнивания по-разному расставили гэпы.
При ручной проверке в Jalview видно, что основные совпадения приходятся на хорошо консервативную центральную часть домена. Различия, напротив, связаны с короткими участками около вставок и гэпов, где последовательностный алгоритм и структурное совмещение могут выбирать немного разные варианты выравнивания.
Визуальный анализ в Jalview согласуется с результатами программы: большая часть колонок совпадает, а основные различия наблюдаются только в коротких участках с гэпами. Это подтверждает корректность программного сравнения.
Обсуждение результатов
Структурное выравнивание и выравнивание ClustalO оказались очень похожими. Из 91 колонки структурного выравнивания 86 колонок совпали с колонками выравнивания ClustalO. Это составляет 94.51% от длины структурного выравнивания. Если считать долю совпадающих колонок относительно длины выравнивания ClustalO, то значение составляет 95.56%.
Такая высокая доля совпадений показывает, что для выбранных трёх белковых структур множественное выравнивание последовательностей хорошо согласуется со структурным выравниванием. Это объясняется тем, что анализировались близкие доменные участки, имеющие сходную пространственную укладку и выраженные консервативные мотивы.
Небольшие различия между двумя выравниваниями связаны преимущественно с короткими участками, содержащими гэпы. Для таких областей возможны разные варианты выравнивания: программа множественного выравнивания ориентируется на сходство последовательностей и штрафы за гэпы, тогда как структурное выравнивание опирается на пространственное расположение остатков в белковой структуре.
Следовательно, в консервативных структурных участках оба подхода дают почти одинаковый результат, а различия проявляются главным образом в вариабельных или плохо совмещаемых фрагментах.
Вывод по заданию 3
В ходе третьего задания было построено выравнивание последовательностей на основе совмещения трёх белковых структур: 2CUB, 4HCK и 1KIK. Также было построено множественное выравнивание тех же последовательностей программой ClustalO. Для визуализации пространственного совмещения было получено изображение structure_superposition.png.
Сравнение структурного выравнивания и ClustalO было выполнено программой compare_alignments.py. Было найдено 86 одинаково выровненных колонок. Они образуют три крупных блока совпадений длиной 29, 25 и 31 колонку, а также одну одиночную совпадающую колонку, не входящую в блок.
Полученные результаты показывают, что выравнивание ClustalO хорошо соответствует структурному выравниванию для выбранных белков. Доля совпадающих колонок составляет 94.51% от длины структурного выравнивания и 95.56% от длины выравнивания ClustalO. Небольшие различия связаны с короткими участками, в которых по-разному расположены гэпы.
Задание 4. Краткое описание программы MSA
Выбранная программа
Для краткого описания была выбрана программа Clustal Omega, так как она использовалась в данной работе при построении множественных выравниваний аминокислотных последовательностей. Clustal Omega относится к программам множественного выравнивания последовательностей и применяется для построения выравниваний белковых, а также нуклеотидных последовательностей.
Назначение программы
Основная задача Clustal Omega состоит в построении множественного выравнивания для трёх и более последовательностей. Такое выравнивание позволяет расположить гомологичные аминокислотные или нуклеотидные остатки в одних и тех же колонках и затем использовать результат для поиска консервативных участков, сравнения белковых семейств, анализа доменной организации и подготовки данных для дальнейших эволюционных или структурных исследований.
В рамках данного практикума Clustal Omega использовалась для выравнивания белковых последовательностей. В задании 2 результат Clustal Omega был принят как выравнивание A и сравнивался с результатами MAFFT и MUSCLE. В задании 3 выравнивание Clustal Omega для последовательностей из PDB-файлов сравнивалось со структурным выравниванием, полученным на основе пространственного совмещения белков.
Общий принцип работы
Clustal Omega строит множественное выравнивание прогрессивным способом. Сначала программа оценивает сходство между последовательностями, затем строит направляющее дерево, которое задаёт порядок объединения последовательностей или групп последовательностей. После этого последовательности постепенно объединяются в общее множественное выравнивание.
Важной особенностью Clustal Omega является использование методов, основанных на скрытых марковских моделях профилей. В отличие от более простого сравнения отдельных последовательностей, профильный подход позволяет учитывать уже накопленную информацию о группе родственных последовательностей. Благодаря этому программа может эффективно работать не только с небольшими наборами данных, но и с большими наборами последовательностей.
Преимущества Clustal Omega
- программа подходит для построения множественных выравниваний белковых последовательностей;
- Clustal Omega достаточно быстро работает даже при сравнительно большом числе последовательностей;
- программа доступна через веб-интерфейс EMBL-EBI, поэтому её можно использовать без установки на локальный компьютер;
- результаты можно скачать в стандартных форматах, например FASTA или Clustal, что удобно для последующего анализа в Jalview и других программах;
- Clustal Omega хорошо подходит для учебных задач, так как имеет понятный веб-интерфейс и может запускаться с настройками по умолчанию.
Ограничения программы
Несмотря на удобство и высокую скорость работы, результат Clustal Omega, как и результат любой программы множественного выравнивания, зависит от выбранного алгоритма и параметров. Наиболее заметные различия между программами обычно возникают в участках с большим числом вставок, делеций и гэпов. Это хорошо видно по результатам задания 2, где выравнивания Clustal Omega, MAFFT и MUSCLE имели разную длину и совпадали только в части колонок.
Кроме того, Clustal Omega строит выравнивание на основе сходства последовательностей, а не напрямую на основе пространственной структуры белков. Поэтому для белков с известными 3D-структурами полезно дополнительно сравнивать последовательностное выравнивание со структурным, как это было сделано в задании 3.
Пример использования в данной работе
В данной работе Clustal Omega применялась двумя способами. Во-первых, с её помощью было построено множественное выравнивание шести белковых последовательностей:
- NCK1_HUMAN/1-377;
- VAV_HUMAN/1-845;
- BLK_MOUSE/1-499;
- FGR_HUMAN/1-529;
- LCK_CHICK/1-508;
- HCK_HUMAN/1-526.
Это выравнивание было сохранено в файл A_clustal.fasta и затем использовалось как выравнивание A при сравнении с MAFFT и MUSCLE.
Во-вторых, Clustal Omega была использована для выравнивания последовательностей из PDB-структур 2CUB, 4HCK и 1KIK. Полученное выравнивание было сохранено в файл ClustalO_pdb_sequences.fasta и затем сравнено со структурным выравниванием. По результатам сравнения было найдено 86 одинаково выровненных колонок из 91 колонки структурного выравнивания, что показывает хорошее соответствие между последовательностным и структурным выравниванием для выбранных белковых доменов.
Источники информации
- EMBL-EBI. Clustal Omega < Multiple Sequence Alignment: https://www.ebi.ac.uk/jdispatcher/msa/clustalo
- Sievers F., Wilm A., Dineen D. et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology, 2011, 7:539. DOI: 10.1038/msb.2011.75.
Вывод по заданию 4
Clustal Omega является удобной и широко используемой программой для множественного выравнивания последовательностей. Она сочетает высокую скорость работы с достаточно хорошим качеством выравнивания и поэтому подходит как для анализа небольших учебных наборов последовательностей, так и для более крупных наборов данных. В данном практикуме Clustal Omega оказалась полезной как базовая программа MSA: её результаты можно было сравнить с другими программами множественного выравнивания, а также со структурным выравниванием белков.