Ресеквенирование. Поиск полиморфизмов у человека.
1. Подготовка чтений:
- Скачал файлы к себе в директорию:
cp /P/y14/term3/block4/SNP/reads/chr22.fastq
cp /nfs/srv/databases/ngs/Human/chr22.fasta - С помощью программы Trimmomatic была произведена очистка чтений (с конца каждого чтения отрезаны нуклеотиды с качеством ниже 20 (TRAILING:20), а также отсеяны чтения длиной меньше 50 нуклеотидов (MINLEN:50):
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chr17.fastq chr17_out.fastq TRAILING:20 MINLEN:50 - С помощью программы FastQC сравнил качество изначального чтения и полученного после очистки:
fastqc chr22.fastq
fastqc chr22_out.fastq
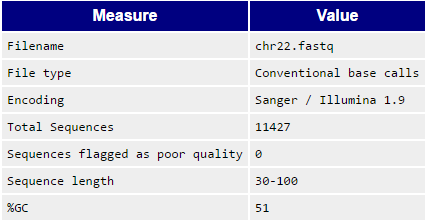
Рисунок 1. Общая статистика до чистки. |
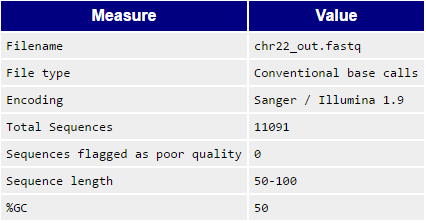
Рисунок 2. Общая статистика после чистки. |
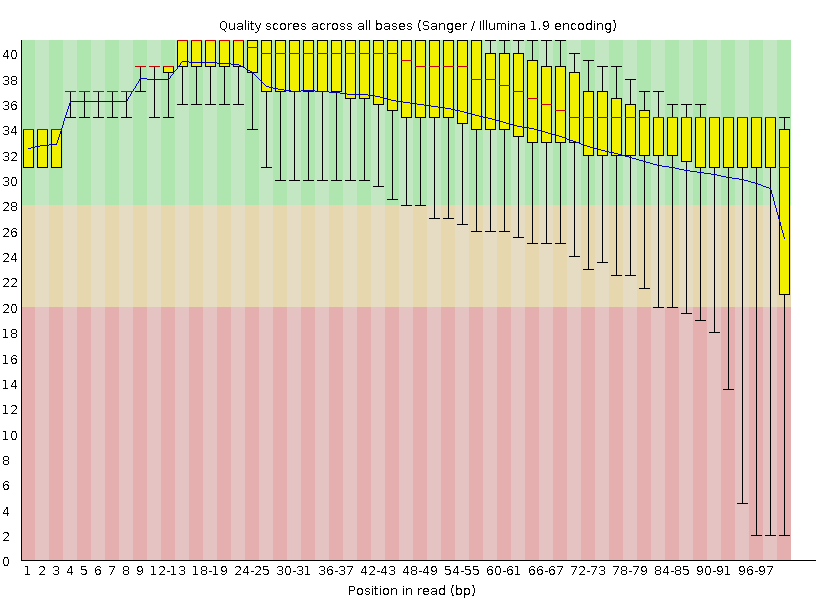
Рисунок 1. "Per base quality" до чистки. |
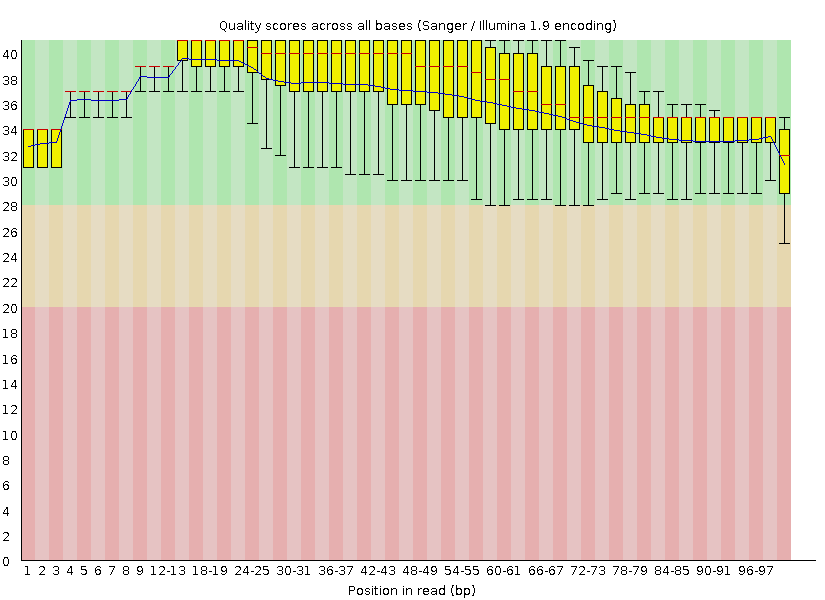
Рисунок 2. "Per base quality" после чистки. |
Отсеялись чтения с длиной меньше 50 нуклеотидов после обрезания низкокачественных концов
2. Картирование чтений:
Для ксанирования очищенных чтений использовалась программа BWA. Сначала я проиндексировал референсную последовательность chr22.fasta:
bwa index chr22.fasta
Затем было построено выравнивание прочтений референса в формате .sam с использованием алгоритма mem:
bwa mem chr22.fasta chr22_out.fastq > align.sam
После этого с помощью пакета samtools файл с выравниванием был переведен в бинарный формат .bam:
samtools view align.sam -b -o align.bam
Получившийся файл был подан на вход другой команде, чтобы отсортировать выравнивание чтений с референсом по координате в референсе начала чтения:
samtools sort align.bam -T tmp.txt -o sort.bam
Параметр -T был задан для записи временных файлов в файл tmp.txt. Далее полученный файл был проиндексирован:
samtools index sort.bam
Затем я выяснил, сколько чтений откартировалось на геном:
samtools idxstats sort.bam > out.out
11090 чтений из 11091 было откартировано на хромосому; таким образом не было картировано только 1 чтение.
3. Анализ SNP:
Сначала был создан файл с полиморфизмами в формате .bcf с помощью команды:
samtools mpileup -uf chr22.fasta sort.bam -o snp.bcf
Потом был создан файл со списком отличий между референсом и чтениями в формате .vcf:
bcftools call -cv snp.bcf -o snp.vcf
Таблица 1. Описание трех полиморфизмов:
| Координата | Тип полиморфизма | В референсе | В ридах | Глубина покрытия | Качество ридов |
|---|---|---|---|---|---|
| 26220887 | Вставка | tcacacacatg | tCACACACATGAAcacacacatg | 2 | 71.224 |
| 26222247 | Замена | T | C | 4 | 90.5135 |
| 28656299 | Делеция | CTATAT | CTAT | 1 | 13.6619 |
Было получено 14 инделей и 214 snp (cat| grep | wc -l). В среднем, качество у найденных полиморфизмов хорошее (около 140), а покрытие не очень (около 20)
Затем необходимо было из файла snp_only.vcf (файл с удаленными инделями) получить файл, с которым может работать программа annovar:
perl /nfs/srv/databases/annovar/convert2annovar.pl -format vcf4 snp-only.vcf -outfile snp.avinput
Далее была произведена аннотация с помощью баз данных: refgene, dbsnp, 1000 genomes, Gwas, Clinvar.
1) База refgene:
perl /nfs/srv/databases/annovar/annotate_variation.pl -out pol.refgene -build hg19 snp.avinput /nfs/srv/databases/annovar/humandb
Были получены файлы pol.refgene.exonic_variant_function(файл с описанием SNP, попавших в экзоны), pol.refgene.log (файл с описанием прохождения работы), pol.refgene.variant_function (файл с описанием SNP по группам).
Таблица 2. Информация о SNP в экзонах
| Координата | Тип замены | Ген | В референсе | В чтениях | Гетеро или гомозиготная замена | Качество чтений | Глубина покрытия |
|---|---|---|---|---|---|---|---|
| 26159289 | несинонимичная | MYO18B | G | A | гомо | 221.999 | 19 |
| 26164390 | синонимичная | MYO18B | G | A | гетеро | 100.008 | 17 |
| 26164855 | несинонимичная | MYO18B | A | T | гетеро | 118.008 | 19 |
| 26166900 | несинонимичная | MYO18B | G | C | гомо | 221.999 | 27 |
| 26173661 | несинонимичная | MYO18B | T | C | гомо | 221.999 | 28 |
| 26222454 | несинонимичная | MYO18B | C | T | гомо | 221.999 | 28 |
| 26239850 | несинонимичная | MYO18B | С | A | гомо | 221.999 | 18 |
| 26286738 | несинонимичная | MYO18B | T | A | гетеро | 225.009 | 62 |
| 26342144 | синонимичная | MYO18B | G | A | гетеро | 75.0075 | 11 |
| 26422980 | несинонимичная | MYO18B | A | G | гетеро | 225.221 | 24 |
| 26423124 | несинонимичная | MYO18B | G | C | гетеро | 180.009 | 19 |
| 26423477 | несинонимичная | MYO18B | G | A | гетеро | 46.0072 | 13 |
| 26423535 | несинонимичная | MYO18B | G | A | гетеро | 46.0072 | 9 |
| 28378450 | несинонимичная | TTC28 | T | C | гетеро | 212.009 | 29 |
| 28378472 | синонимичная | TTC28 | A | G | гомо | 222.01 | 34 |
| 28389453 | синонимичная | TTC28 | T | C | гетеро | 51.0072 | 15 |
| 28504183 | синонимичная | TTC28 | G | A | гетеро | 167.009 | 17 |
| 28504258 | синонимичная | TTC28 | A | G | гетеро | 154.008 | 17 |
| 36661330 | несинонимичная | APOL1 | G | A | гетеро | 175.009 | 34 |
| 36661536 | синонимичная | APOL1 | C | A | гомо | 221.999 | 19 |
| 36661566 | несинонимичная | APOL1 | G | A | гомо | 221.999 | 17 |
| 36661646 | несинонимичная | APOL1 | G | A | гомо | 221.999 | 18 |
| 36661674 | несинонимичная | APOL1 | C | A | гетеро | 155.008 | 20 |
| 36661842 | синонимичная | APOL1 | G | A | гомо | 221.999 | 52 |
| 36661906 | несинонимичная | APOL1 | A | G | гетеро | 225.009 | 72 |
| 36662034 | несинонимичная | APOL1 | T | G | гетеро | 196.009 | 36 |
Таблица 3. Информация о SNP в различных группах
| В интронах | В экзонах | ncRNA_intronic | ncRNA_exonic | Гомо | Гетеро |
|---|---|---|---|---|---|
| 181 | 26 | 6 | 1 | 139 | 75 |
Из Таблицы 3 следует, что больше всего SNP в интронах, так как замена в некодирующей последовательности не приводит к замене аминокислоты. SNP в экзонах были лишь в генах MYO18B, TTC28 и APOL1.
2) База dbsnp:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out pol.dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb
Были получены файлы pol.dbsnp.hg19_snp138_dropped (SNP c rs), pol.dbsnp.hg19_snp138_filtered (без rs), pol.dbsnp.log. 176 SNP имеют rs, 38 нет.
3) База 1000 genomes:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -dbtype 1000g2014oct_all -buildver hg19 -out pol.1000 snp.avinput /nfs/srv/databases/annovar/humandb
Были получены файлы pol.1000.hg19_ALL.sites.2014_10_dropped, pol.1000.hg19_ALL.sites.2014_10_filtered, pol.1000.log.
Состав файлов здесь такой же, как в прошлой базе данных. Однако здесь аннотировано лишь 171, а 43 нет. Среднее значение частоты 0.374632128, минимальное - 0.00319489, максимальное - 0.9998.
4) База Gwas:
perl /nfs/srv/databases/annovar/annotate_variation.pl -regionanno -build hg19 -out pol.gwas -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb
Были получены файлы pol.gwas.hg19_gwasCatalog, pol.gwas.log. В основном файле содежатся SNP, имеющие какое-то значение в медицине. В моем случае 3 SNP аннотированы в данной базе данных. Один связан с математическими способностями детей с дислексией, другой с ожирением, а третий с гломерулосклерозом.
5) База Clinvar:
perl /nfs/srv/databases/annovar/annotate_variation.pl -filter -out pol.clinvar -dbtype clinvar_20150629 -buildver hg19 snp.avinput /nfs/srv/databases/annovar/humandb
Были получены файлы clinvar.hg19_clinvar_20150629_dropped, clinvar.hg19_clinvar_20150629_filtered, pol.clinvar.log. Аннотированы 2 SNP в генах, связанных с фокальным сегментарным гломерулосклерозом.