Поиск сигналов. chip-seq
1. Контроль качества прочтений
Я работал с файлом chipseq_chunk53.fastq с ридом Illumina, полученный в результате сhip-seq эксперимента, соответствующий участку хромосомы.
Сначала был проведен контроль качества прочтений с помощью программы FastQC:
fastqc chipseq_chunk53.fastq
В выходном файле программы можно заметить, что во многих позициях были выпадения по качеству ниже 20 (Рис. 1), поэтому результаты были дальше отфильрованы программой Trimmomatic (с конца каждого чтения отрезаны нуклеотиды с качеством ниже 50 (TRAILING:50), а также отсеяны чтения длиной меньше 36 нуклеотидов (MINLEN:36)):
java -jar /usr/share/java/trimmomatic.jar SE -phred33 chipseq_chunk53.fastq trimm.fastq TRAILING:50 MINLEN:36
Затем с помощью FastQC снова был проведен контроль качества прочтений. Изначально было 16622 ридов, а после очистки их осталось 14174 (85.27%); также средние значения качества улучшились, и нету выпадений ниже 25 (Рис. 2).
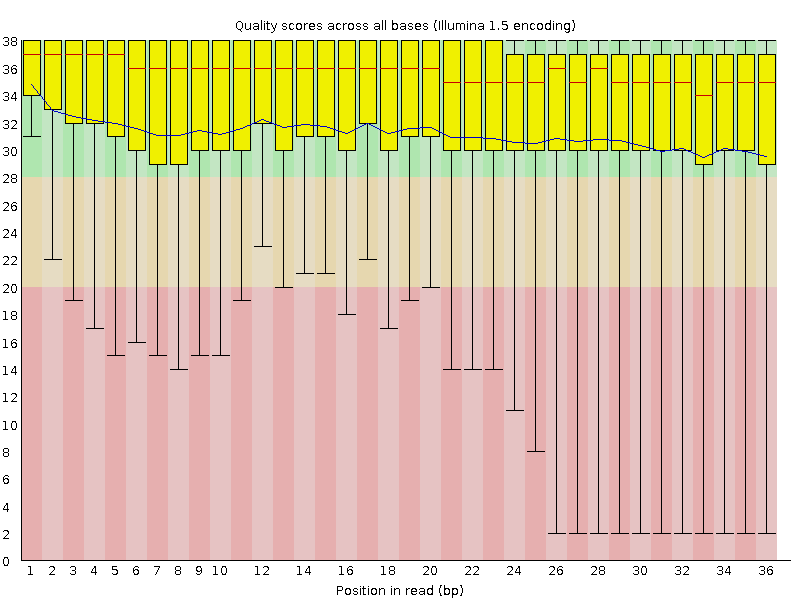
Рисунок 1. Per base quality до очистки.
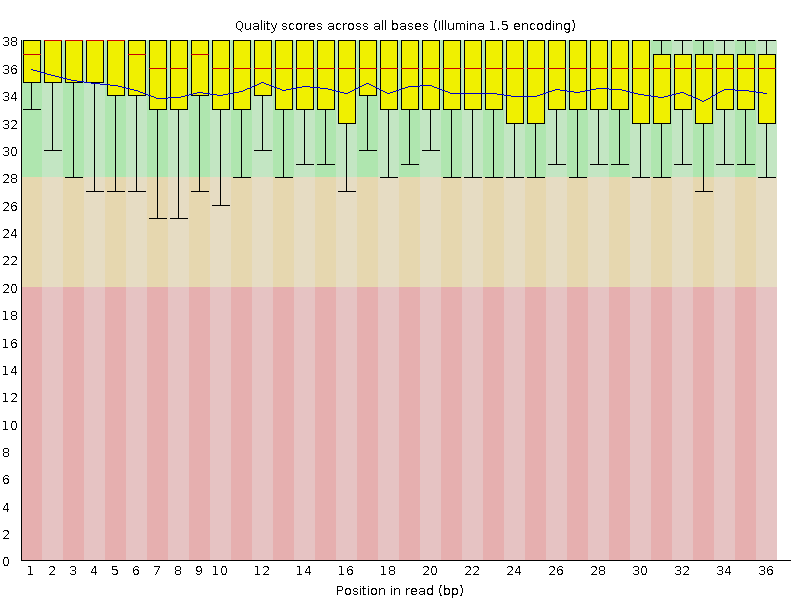
Рисунок 2. Per base quality после очистки.
2. Картирование прочтений на геном человека hg19
Картирование прочтений на геноме человека hg19 осуществлялось с помощью программы BWA:
bwa mem /srv/databases/ngs/hg19/GRCh37.p13.genome.fa chipseq_chunk53.fastq > chipseq_chunk53.sam
Затем полученные данные были проанализированны:
samtools view -bSo chipseq_chunk53.bam chipseq_chunk53.sam - файл с выравниванием переводится в бинарный формат .bam
samtools sort chipseq_chunk53.bam -T chip_temp -o chipseq_chunk53.sorted.bam - сортирует выравнивание ридов и референса по координате рида в референсе
samtools index chipseq_chunk53.sorted.bam - полученный файл сортируется
samtools idxstats chipseq_chunk53.sorted.bam > chipseq_chunk53.idxstats - считает, сколько ридов откартировалось
В результате был получен файл с откартированными на геном ридами. Откартировались все 16622 ридов, 14778 (88.91%) из которых откартировались на первую хромосому. Таким образом, скорее всего, для анализа мне были предложены прочтения с первой хромосомы.
3. Поиск пиков
Для поиска пиков использовалась программа MACS
macs2 callpeak -n chipseq_chunk53 -t chipseq_chunk53.sorted.bam --nomodel
В результате было получено 3 файла: chipseq_chunk53_summits.bed, chipseq_chunk53_peaks.xls, chipseq_chunk53_peaks.narrowPeak. Всего было найдено 12 пиков, все в первой хромосоме. Их визуалиция в UCSC Genome Browser показана на Рис. 3. Для визуализации в браузер загружался файл chipseq_chunk53_peaks.narrowPeak с дописаной в начало строкой - "track type=narrowPeak visibility=3 db=hg19 name="my_peaks" description="Peaks from chunk 53" browser position chr1:9173134-10659851".

Рисунок 3. Визуалиция пиков в UCSC Genome Browser.
Таблица 1. Характеристики пиков.
| Номер | Координаты | Длина | Расстояние от старта пика до его вершины | -log(p-value) |
|---|---|---|---|---|
| 1 | 9173135 - 9173490 | 356 | 164 | 28.15574 |
| 2 | 9232273 - 9232472 | 200 | 27 | 14.57349 |
| 3 | 9254185 - 9254505 | 321 | 136 | 23.03367 |
| 4 | 9495326 - 9495549 | 224 | 93 | 29.37207 |
| 5 | 9598012 - 9598332 | 321 | 130 | 42.0782 |
| 6 | 9610315 - 9610543 | 229 | 162 | 16.35752 |
| 7 | 9613786 - 9614201 | 416 | 306 | 32.42684 |
| 8 | 9974388 - 9974720 | 333 | 192 | 32.65349 |
| 9 | 10116248 - 10116615 | 368 | 220 | 30.46644 |
| 10 | 10231216 - 10231468 | 253 | 95 | 18.7966 |
| 11 | 10549903 - 10550226 | 324 | 175 | 21.74618 |
| 12 | 10659649 - 10659851 | 203 | 26 | 15.94685 |
Каждому найденному пику соответствует число — -log10(p-value), по которому можно оценить достоверность находки. Чем больше это число, тем меньше p-value, и, следовательно, тем выше достоверность находки. Рассмотрим в более крупном масштабе пики с наименьшим p-value, т.е. пики с номерами 2 и 12 в Таблице 1.

Рисунок 4. Пик 2 в более крупном масштабе.

Рисунок 5. Пик 12 в более крупном масштабе.
Второй пик пересекается с геном mir-34 (Рис. 4). Молекулы группы mir-34 microRNA представлены малыми некодирующими РНК (мкРНК), которые у млекопитающих определяют группу, состоящую из трёх зрелых форм малых интерферирующих РНК (миРНК). Двенадцатый пик пересекается с геном PEX14 (Рис. 5), кодирующим одноименный мембранный белок пероксисом у человека.