Ход работы
- В директории /nfs/srv/databases/ngs/ создана директория и туда скопирован файл с 3 хромосомой и одноконцевые чтения
- Затем референс был проиндексирован: hisat2-build chr3.fasta index3
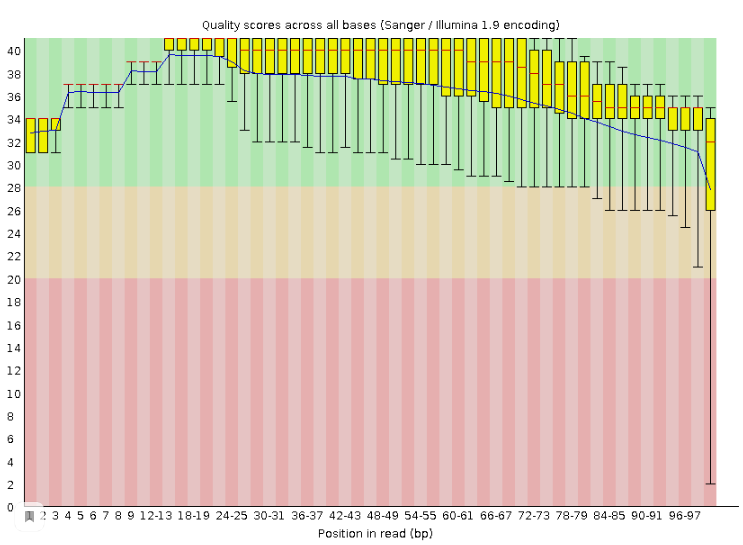
- Далее делаем контроль качества наших прочтений: fastqc chr3.fastq (Рис.1)
- Производим чистку наших ридов: java -jar /nfs/srv/databases/ngs/suvorova/trimmomatic/trimmomatic-0.30.jar SE -phred33 chr3.fastq chr3trim1.fastq TRAILING:20 MINLEN:50
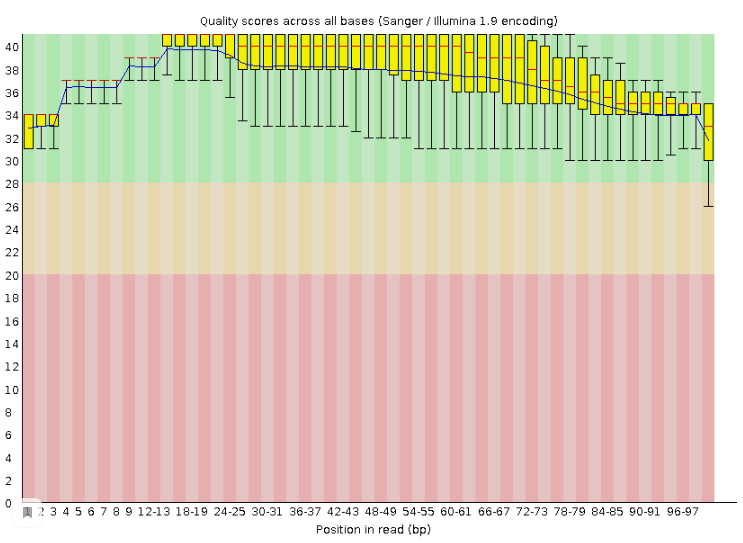
- Производим контроль качества очищенных ридов: fastqc chr3trim1.fastq (Рис.2)
- Изначально чтений было 20932 после очистки стало 20570
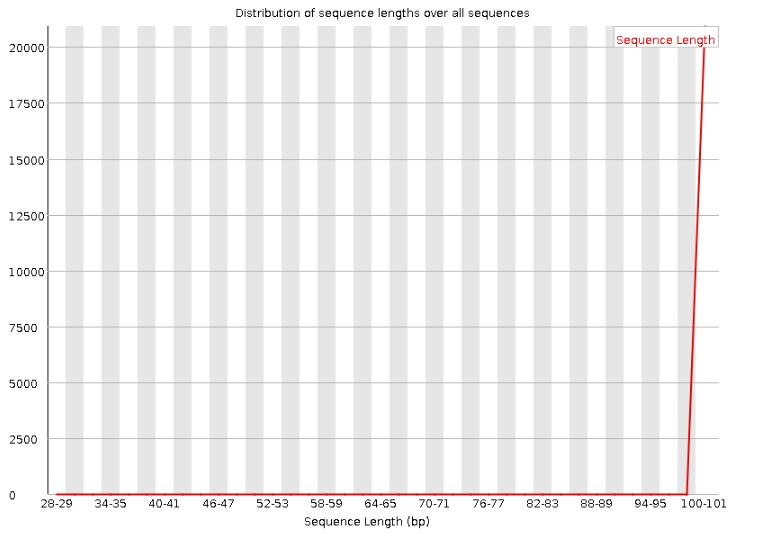
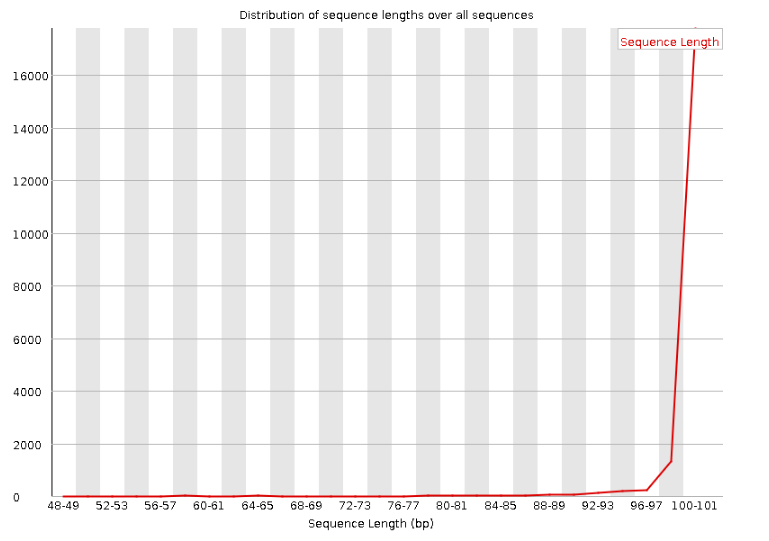
- Очистка чтений оказалась довольно полезной. Заметно уменьшились планки разброса quality, а также увеличился средний показатель quality для последней четверти длины чтения. График длин чтений стал более пологим (т.к. прочтения длины 100 потеряли нуклеотиды на конце и стали более короткими). А так же мы избавились от чтений короче 50 нуклеотидов. (Рис. 3 и 4)
- Добавляем пакет программ в path (чтобы можно юыло вызывать их через командную строку) : PATH=${PATH}:/home/students/y06/anastaisha_w/hisat2-2.0.5
- Создаем выравнивания референса с прочтениями: hisat2 -x index3 -U chr3trim1.fastq -S chr3align.sam --no-spliced-alignment --no-softclip
- Переводим выравнивания в бинарный формат: samtools view -b chr3align.sam -o chr3align.bam
- Сортируем бинарное выравнивание: samtools sort chr3align.bam chr3alignsorted
- Индексируем отсортированное бинарное выравнивание: samtools index chr3alignsorted.bam
- Выдача запроса (samtools flagstat chr3alignsorted.bam)
20589 + 0 in total (QC-passed reads + QC-failed reads) 19 + 0 secondary 0 + 0 supplementary 0 + 0 duplicates 20510 + 0 mapped (99.62%:-nan%) 0 + 0 paired in sequencing 0 + 0 read1 0 + 0 read2 0 + 0 properly paired (-nan%:-nan%) 0 + 0 with itself and mate mapped 0 + 0 singletons (-nan%:-nan%) 0 + 0 with mate mapped to a different chr 0 + 0 with mate mapped to a different chr (mapQ>=5)
- Видно, что 20510 прочтений всего легло на на референс, из которых 19 дважды. Из этого можно сделать вывод о хоошем качестве картирования
- Создаем файл с полиморфизмами: samtools mpileup -u -f chr3.fasta -o snp.bcf chr3alignsorted.bam
- Переводим бинарный файл в читаемый: bcftools call -cv -o snp.vcf snp.bcf (данные в Таб.1)
- Избавляемся от инделей: vcftools --vcf snp.vcf --remove-indels --recode --out snp_noin
- Всего было найдено 230 полиморфизмов, из которых 218 - SNP, 41 не имеют rs, инделей 12
- Конвертируем: convert2annovar.pl -format vcf4 snp_noin.recode.vcf -out snp.avinput
- dbsnp: annotate_variation.pl -filter -out dbsnp -build hg19 -dbtype snp138 snp.avinput /nfs/srv/databases/annovar/humandb.old/
- refgene: annotate_variation.pl -out refgene -build hg19 -dbtype refGene snp.avinput /nfs/srv/databases/annovar/humandb.old/
- RefSeq в annovar разделяет снипы по положению (мне он выделил 13 exonic снипов)
- Также аннотация RefSeq позволяет судить о нуклеотидных и аминокислотных заменах:
line49 nonsynonymous SNV ULK4:NM_017886:exon33:c.A3292G:p.K1098E, chr3 41504679 41504679 T C het 88.0076 .
После c указаны замещаемое основания, позиция, замещающее основание. После p указаны замещаемая аминокслота, позиция, замещающая аминокислота. Видно в данном примере, что замена не синонимичная - Большинство SNP попали в гены: GNL3, FNDC3B, ULK4
- 1000 genomes: annotate_variation.pl -filter -out 1000g -buildver hg19 -dbtype 1000g2014oct_all snp.avinput /nfs/srv/databases/annovar/humandb.old/
- Gwas: annotate_variation.pl -regionanno -out gwas -build hg19 -dbtype gwasCatalog snp.avinput /nfs/srv/databases/annovar/humandb.old/
- В Gwascatalog изложена информация о 4 SNP: ULK4-кровяное давление; GNL3 - уровень аденопектина; 2 SNPs FNDC3B - рост
- Clinvar: annotate_variation.pl -filter -out clinvar -buildver hg19 -dbtype clinvar_20150629 snp.avinput /nfs/srv/databases/annovar/humandb.old/
- Clinvar данных не дал (dropped пустой)
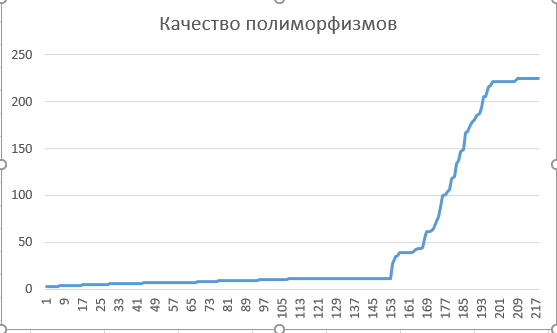
- Так же был построен график качетва от номера полиморфизма (при этом среднее значение = 48.56) (Рис.5)
| Координаты | Тип полиморфизма | Изменение | Глубина | Качество |
| 41291081 | Замена | G -> A | 25 | 221.9 |
| 171897685 | Вставка | cagg -> cAGGagg | INDEL | 22.5 |
| 41359534 | Замена | A -> G | 1 | 6.98 |