В ней представлен внеклеточный домен человеческой карбоангидразы 12.
Карбоангидразы (CA) - ферменты, катализирующие обратимую гидратацию диоксида углерода. Основная функция
этого семейства ферментов это поддержание кислотно-основного баланса, транспорт гидрокарбоната и выработка некоторых
биологических жидкостей. Все представители этого семейтва являются цинковыми металлоферментами. У человека известны как
минимум 13 ферментов этого семейства с различными структурами, функциями и тканеспецифичностью.
Интерес к этому семейству наблюдается из-за найденной для отдельных ферментов (CA II , IX и XII) оверэкспрессии в
некоторых злокачественных опухолях.
Получение данной структуры вместе с ингибитором - ацетазоламидом направлено как раз на изучение структуры как самого
фермента так и его взаимодействия с ингибитором для дальнейшего использования этой информации в дезайне молекул с
противоопухолевыми свойствами (статья).
Сама структура имеет наилучшее разрешение 1.5 Å при покрытии интервала рефлексов
40 - 1.5 Å 94.8% и 1.5 Å при покрытии интервала рефлексов 37.93 - 1.50 94.7%.
Мне кажется, что это довольно неплохие показатели.
Моя запись содержит в себе 4 уникальных типа молекул (сам фермент, ион цинка, лиганд-ацетазоламид и вода), всего
4699 атомов, среди которых нет ниодного изотопа водорода. В структуре отсутствуют атомы с нулевой заселенностью
и также отсутствуют аминокислотные остатки с альтернативными конформациями атомов.
По результатам валидации значение R-фактора для модели 0.19 и Rfree 0.207, что говорит о достаточно хорошем
соответствии модели экспериментально полученным моделям структурных факторов; Rfree - R < 10%, скорее всего модель не
переоптимизирована.
В структуре отсутствуют аутлайеры по длине, углам, хиральности и планарности ковалентных связей.
Всего в структуре определены 50 клэшей с учетом добавленных водородов, в среднем 6 клэшей на 1000 атомов.
В цепи А проанализированы 258 аминокислот из 263 на маргинальность по углам Ψ и Φ, при этом 0 аутлайеров, а 7 а.о.
отнесены к категории допустимых. В цепи B проанализированы 257 аминокислот из 263 на маргинальность по углам Ψ и Φ,
при этом 1 аутлайер, а 5 а.о. отнесены к категории допустимых.
Относительно типичных торсионных углов боковых цепей (ротамеров)
маргинальными являются 0.4% остатков (2 аутлайера) при проанализированных 232 и 231 (цепь A и B соответственно), что является
хорошим результатом.
RSR Z-score выше 2 у 4.8% аминокислотных остатков, что кажется довольно плохим показателем.
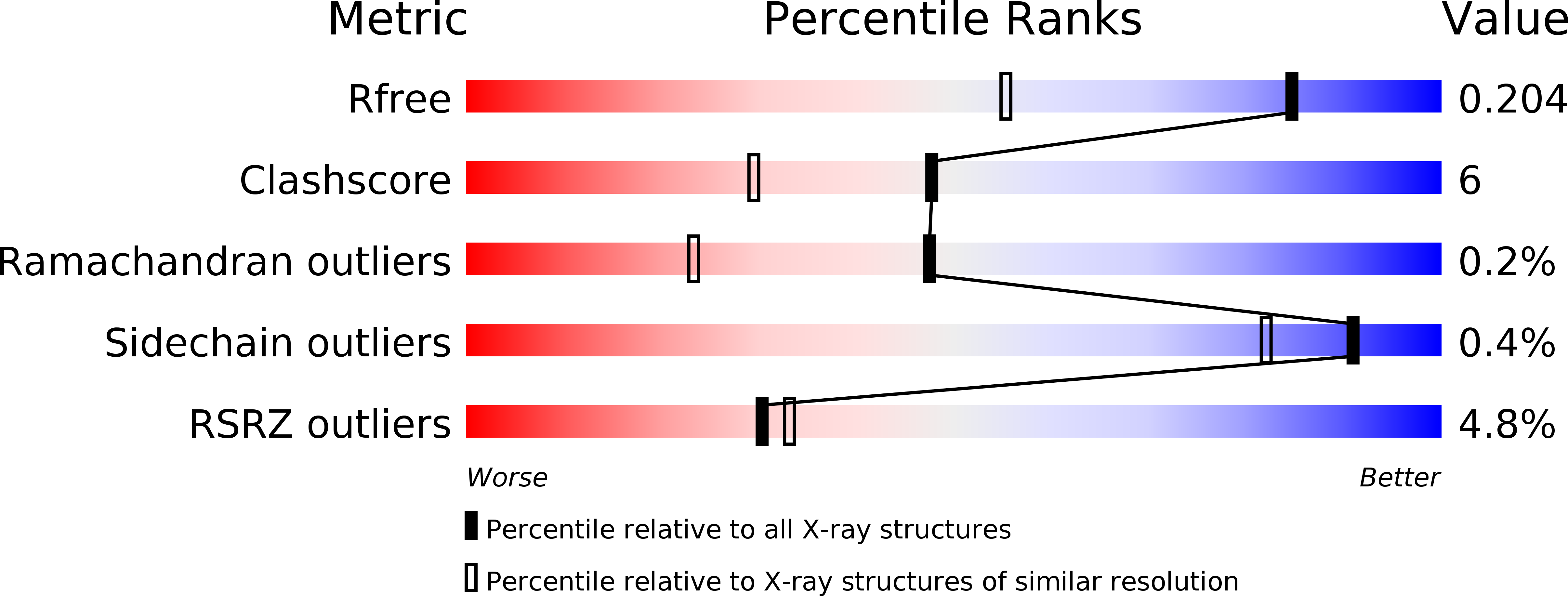
На Рисунке 1 показано соотношение между метриками данной модели и всех других в банке PDB. Особенно
выделяется высокий показатель RSRZ > 2. При этом среднее значение RSRZ по а.о. 0.31. Кажется, что хоть количество
аутлайеров довольно большое, но остальные а.о. хорошо соответствуют экспериментальным данным, о чем еще может говорить
довольно хорошие показатели R и R free.
Рис 1. Параметры качества модели относительно банка PDB.
На Рисунке 2 представлено сопоставление качества разных цепей. Зеленым показан
процент остатков, не являющихся маргиналами по геометрическим критериям, желтым -
являющимися по одному критерию, оранжевым - по 2, красным - по 3 или больше. Серым указана
доля остатков, не представленных в модели. Красным сверху -
процент остатков, плохо вписывающихся в свою ЭП. Видно, что цепи сильно похожи
между собой по этим параметрам.
Рис 2. Качество белковых цепей в структуре.
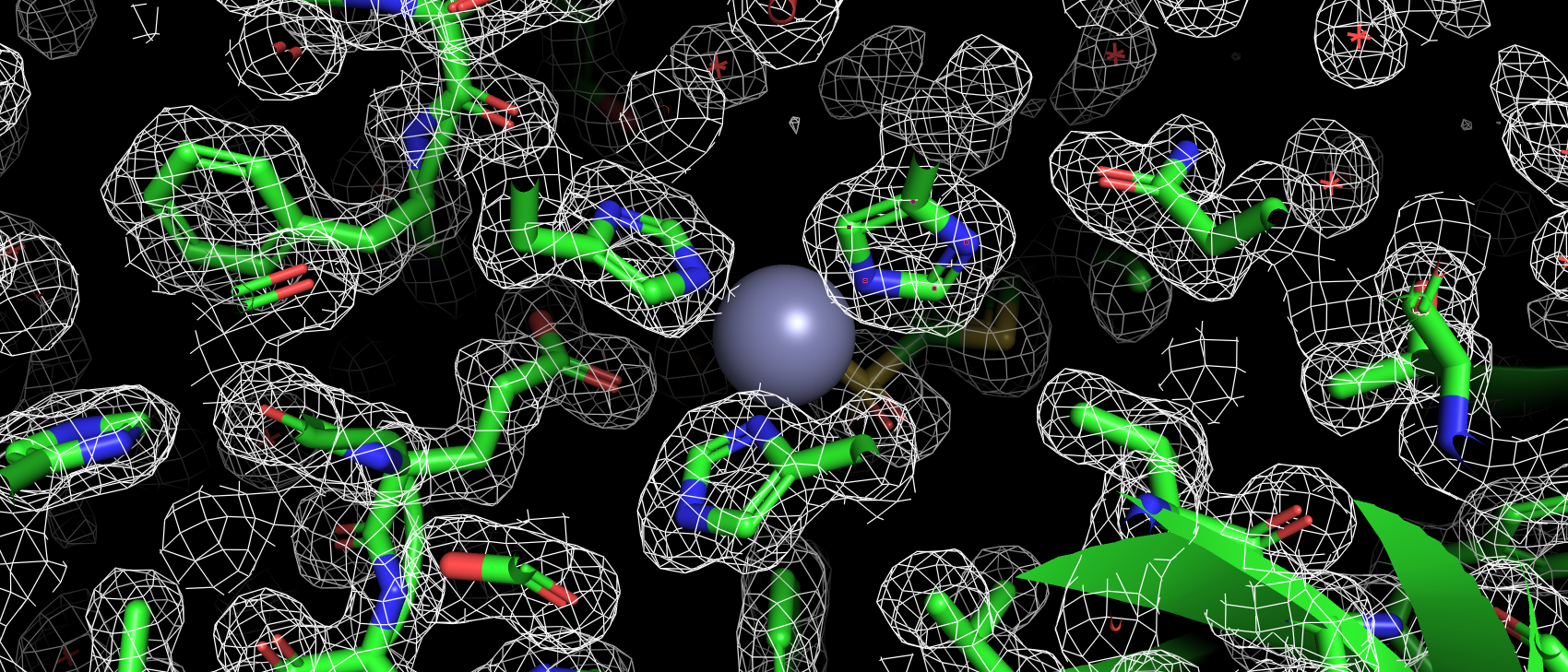
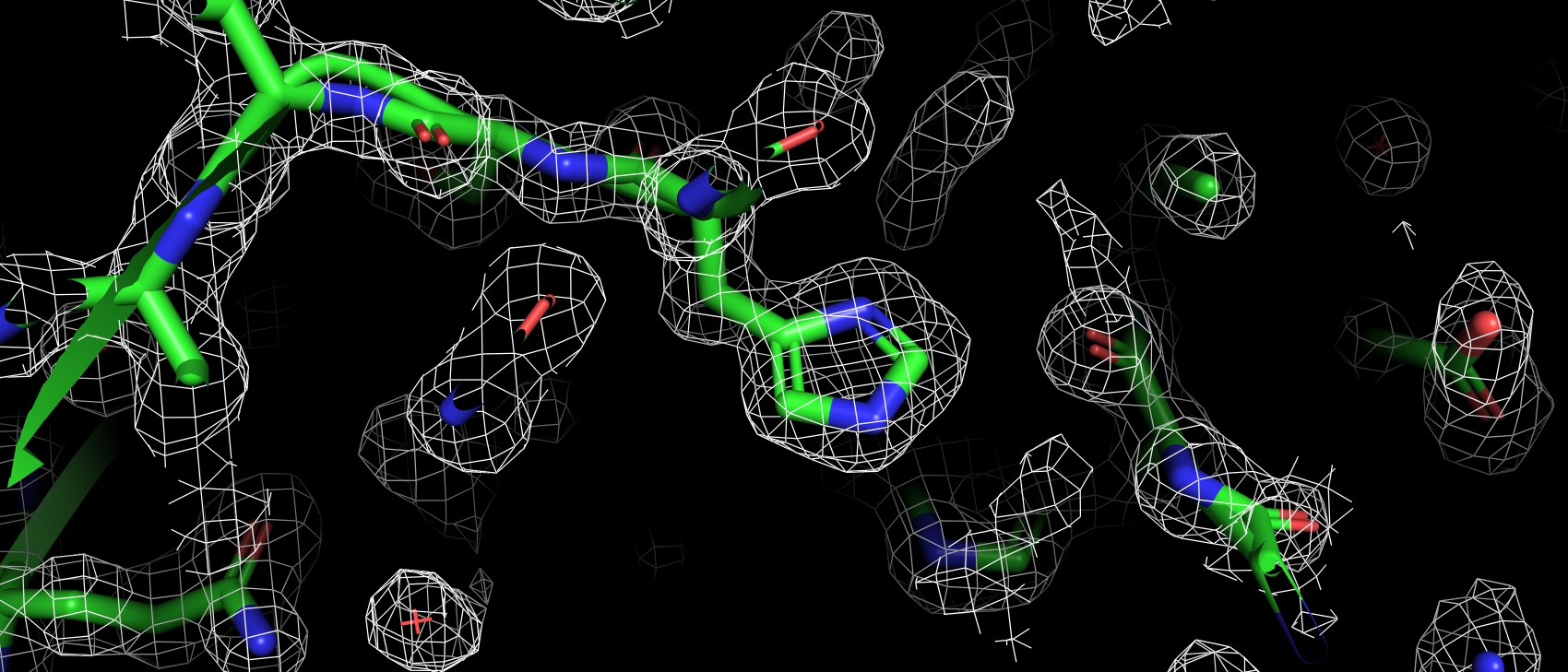
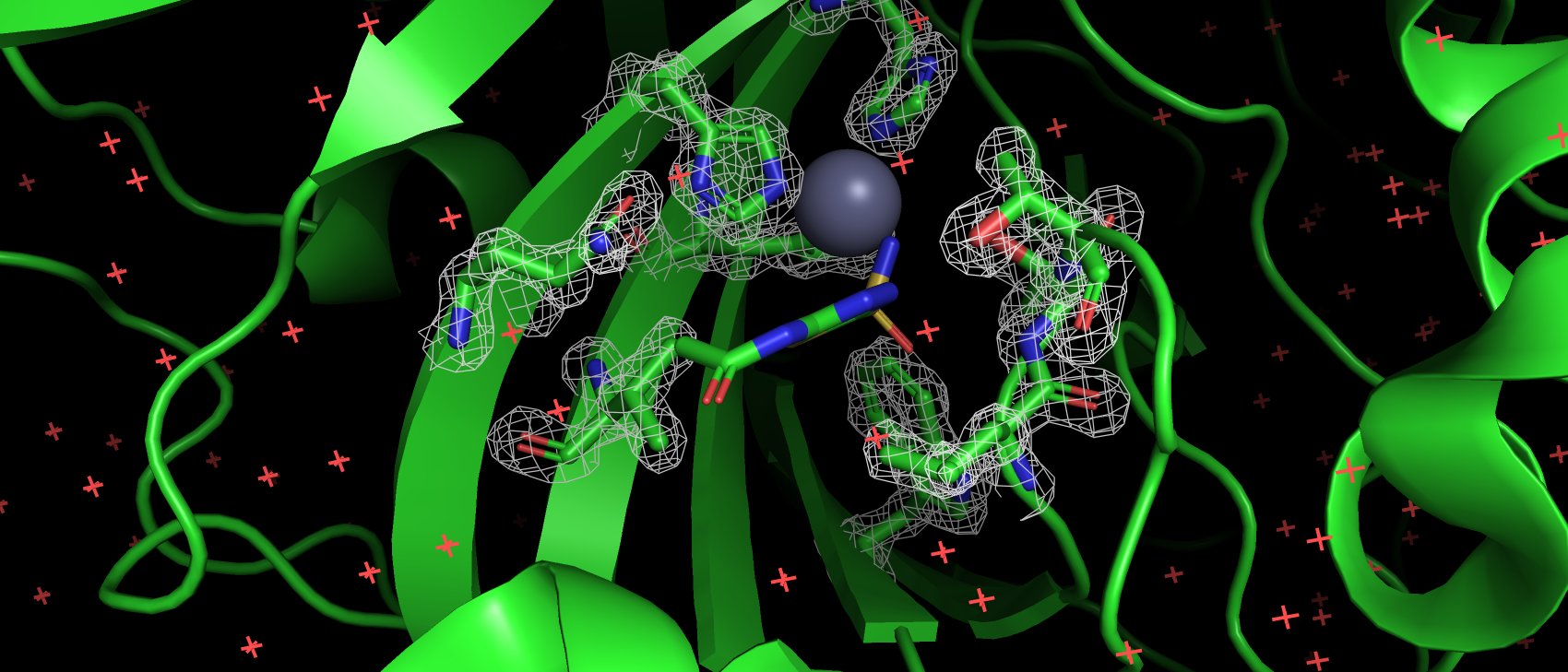
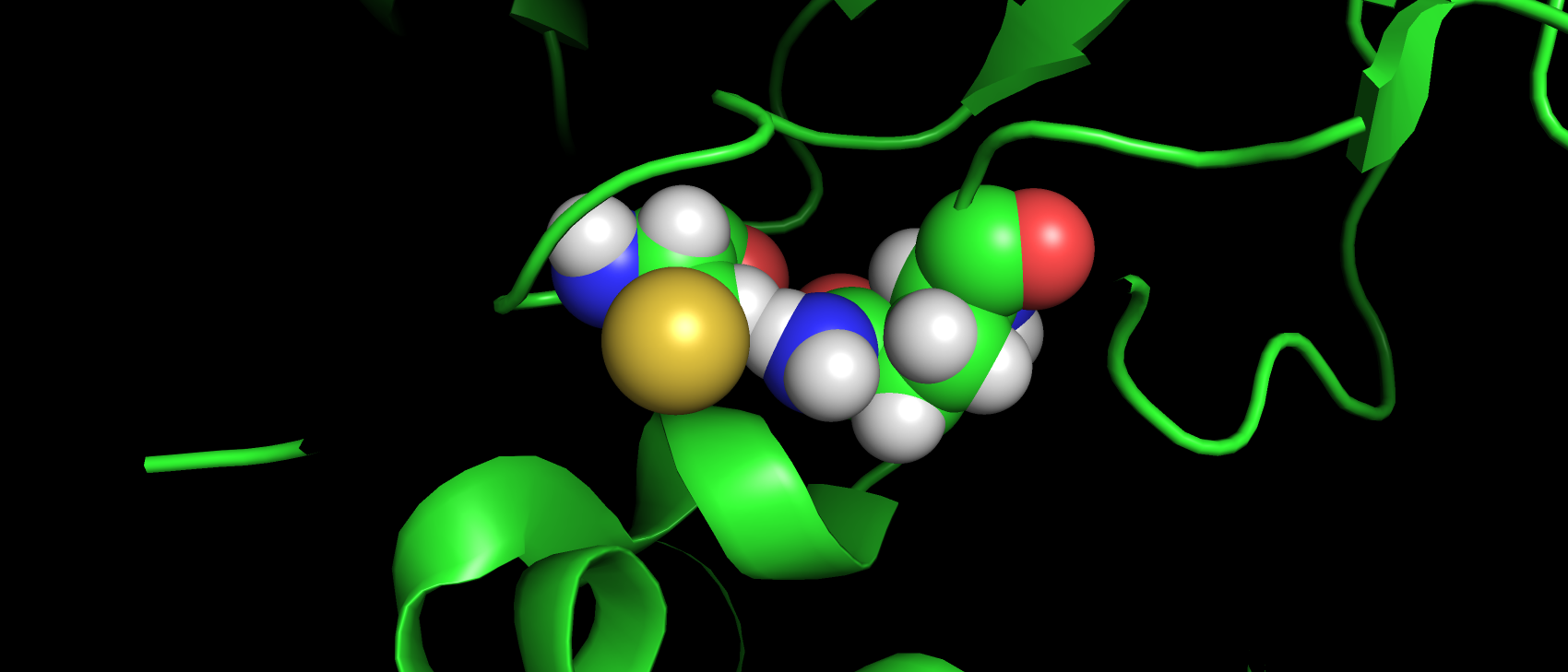
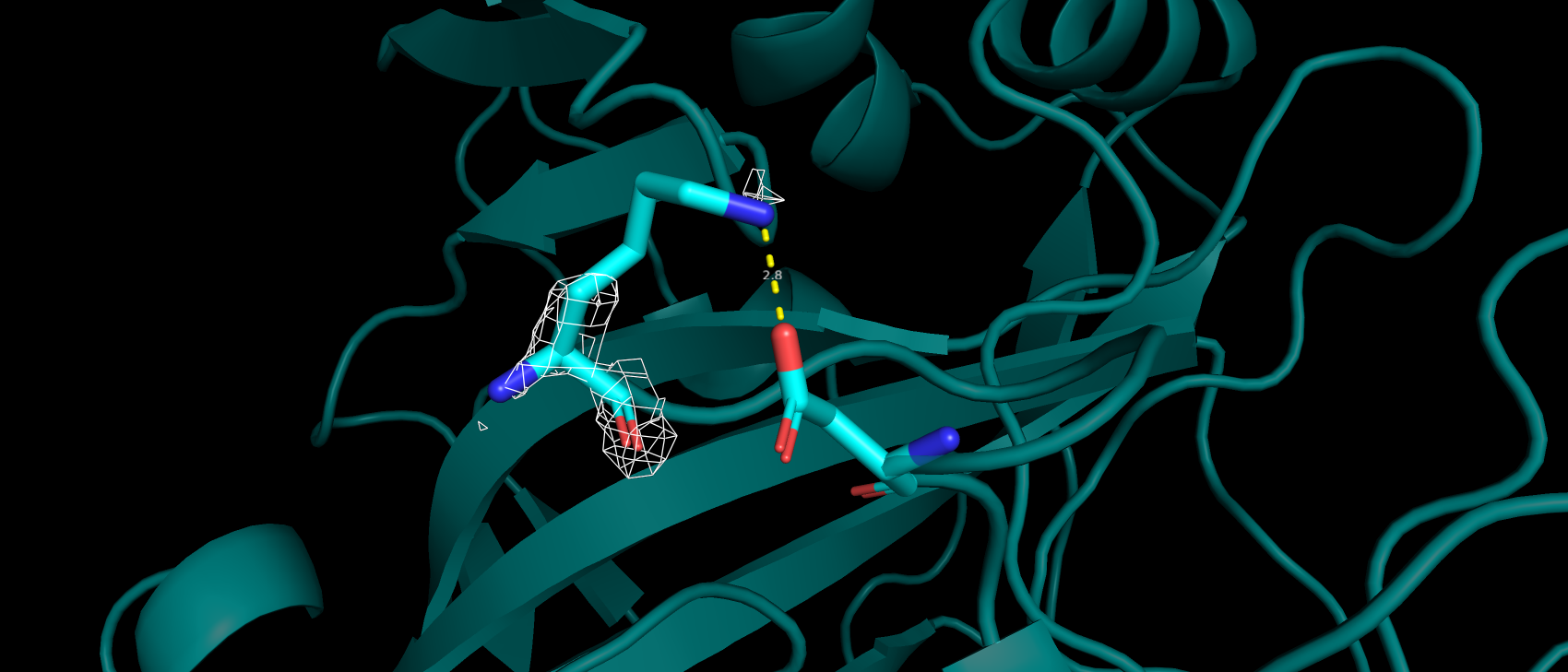
В базе данных Uniprot (O43570) только 4 аминокислотных остатка имеют аннотированную функцию: 3 связывают ион цинка
(119, 121 и 145), 1 приурочен к активному центру как донор протона (94). В структуре pdb эти а.о. имеют номера: 94,
96, 119 и 64 соответственно. Так же так как эта структура направлена на изучение взаимодействия с лигандом важно, чтобы
аминокислоты вступающие с ним во взаимодействия тоже не являлись аутлайерами по какому-либо критерию. Сайт связывания
ацетазоламида: 92, 94, 96, 119, 121, 198, 199, 200 и 209 (нумерация по pdb). Получилось, что ниодна из этих аминокислот
не является аутлайером по какому-либо критерию. Более того каждая из них имеет подтверждающую их ЭП.
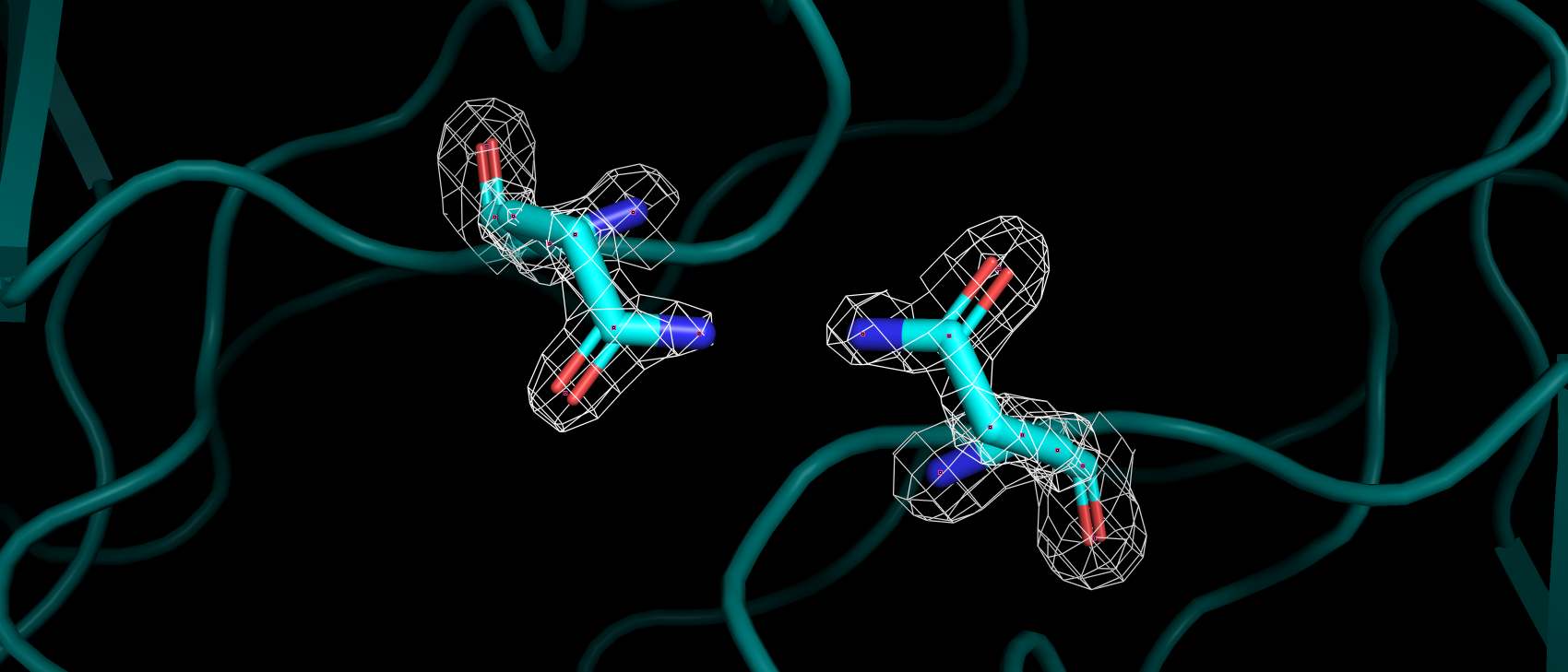
Рис 3. ЭП 94, 96 и 119 гистидинов, участвующих в связывании иона цинка.
Рис 4. ЭП 64 гистидина, аннотированного как АЦ.
Рис 5. ЭП а.о., аннотированных как сайт связывания лиганда-ацетазоламида.
Задание 2
Первыми маргиналами я выбрал два а.о. A:28:GLN и A:203:CYS, которые входят в список слишком близких контактов.
Для них указано пересечение Ван-дер-Вальсовых радиусов атомов водорода, что отображено на рис.6.
Рис 6. Перекрывание Ван-дер-Вальсовых радиусов атомов водорода A:28:GLN и A:203:CYS.
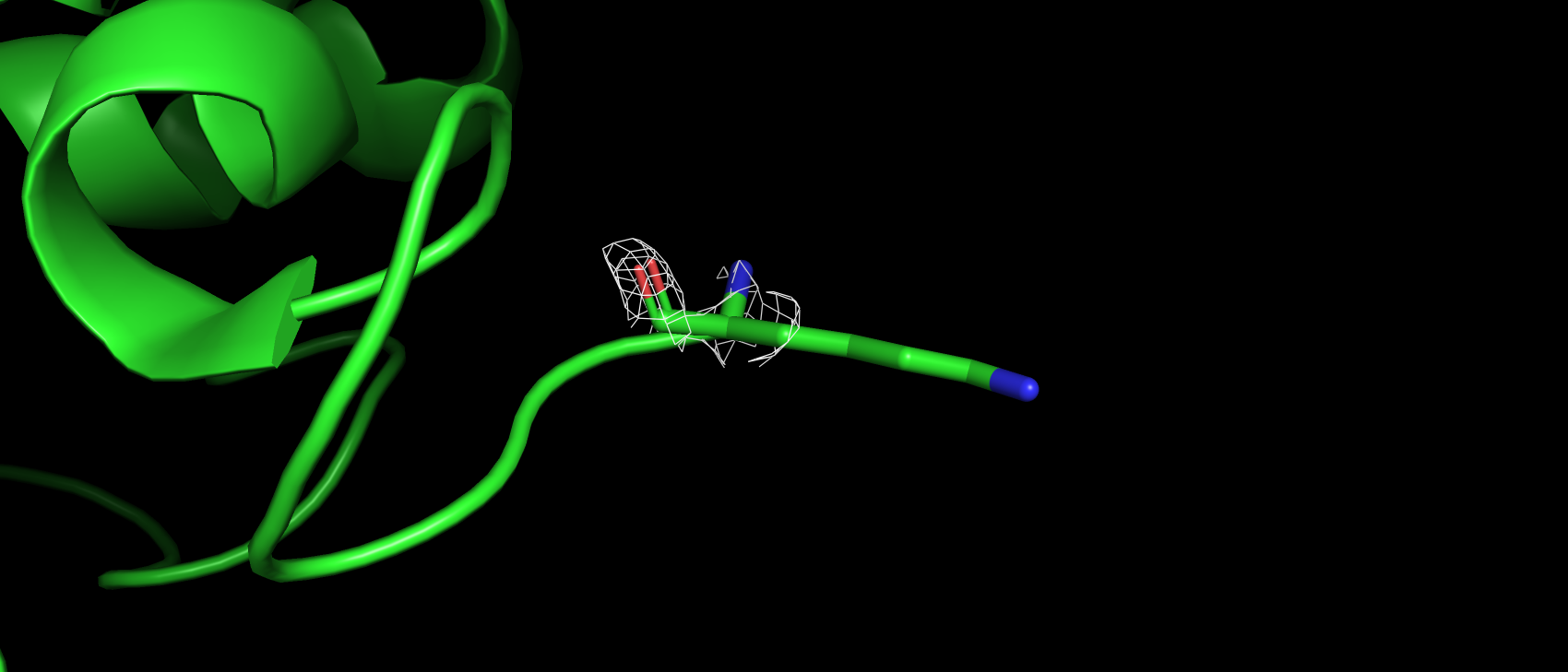
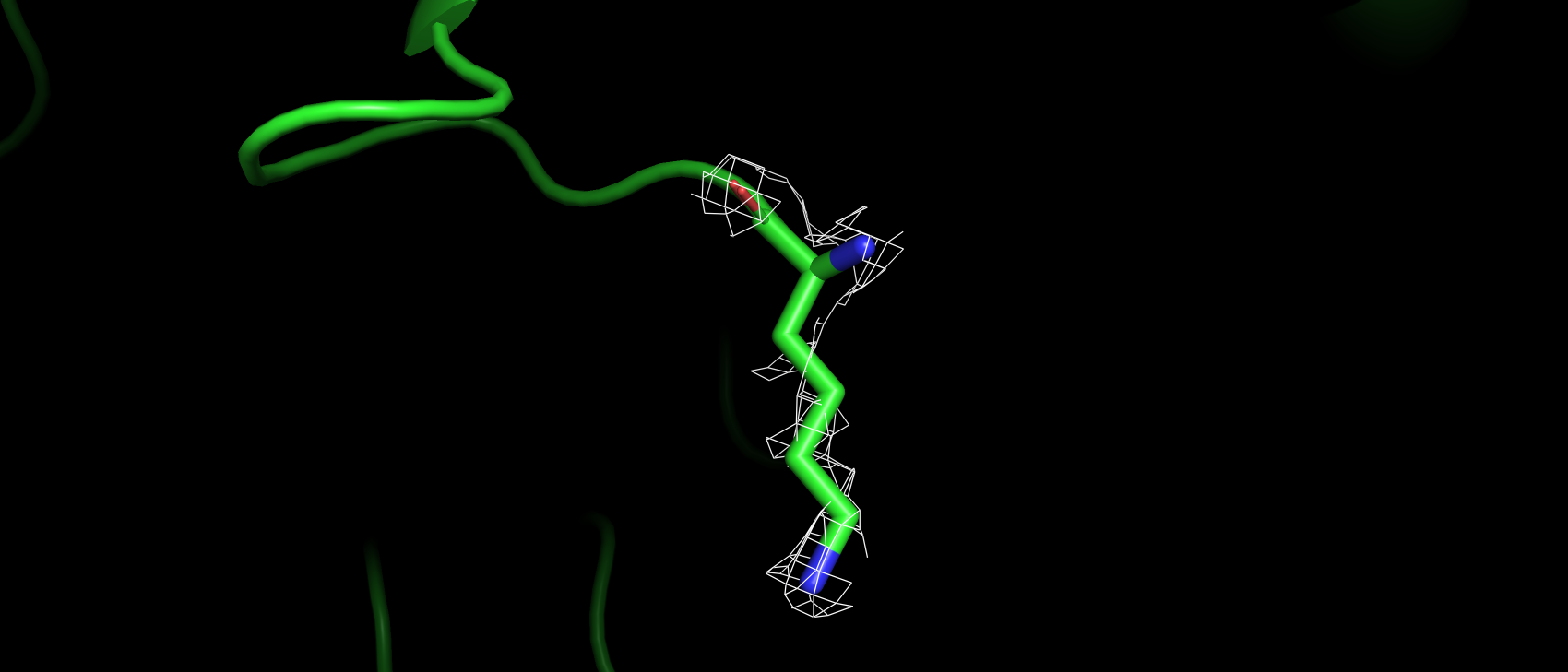
Вторым маргинальным остатком стал A:4:LYS, по показателю RSRZ, который для него составил 6.4. При попытке
визуализировать ЭП вокруг него, только на уровне подрезки 1 появляются намеки на ЭП (рис. 7), которое хотя бы частично
окружает атомы аминокислотного остатка. При еще более низких уровнях подрезки ЭП все также отрывками покрывает отдельные
атомы (рис. 8). Это может объясняться тем, что данный остаток находится на самой поверхности белковой молекулы
и как подвижен в растворе, так и подвержен тепловым колебаниям в кристалле.
Рис 7. ЭП на уровне подрезки 1 а.о. A:4:LYS.
Рис 8. ЭП на уровне подрезки 0.25 а.о. A:4:LYS.
В качестве третьего примера маргинальности я выбрал еще два остатка, которые входят в список слишком близких контактов, но теперь
наблюдается пересечение Ван-дер-Вальсовых радиусов атомов азота A:112:GLN:NE2 и B:112:GLN:NE2 (рис.9).
Рис 9. Перекрывание Ван-дер-Вальсовых радиусов атомов азота A:112:GLN:NE2 и B:112:GLN:NE2.
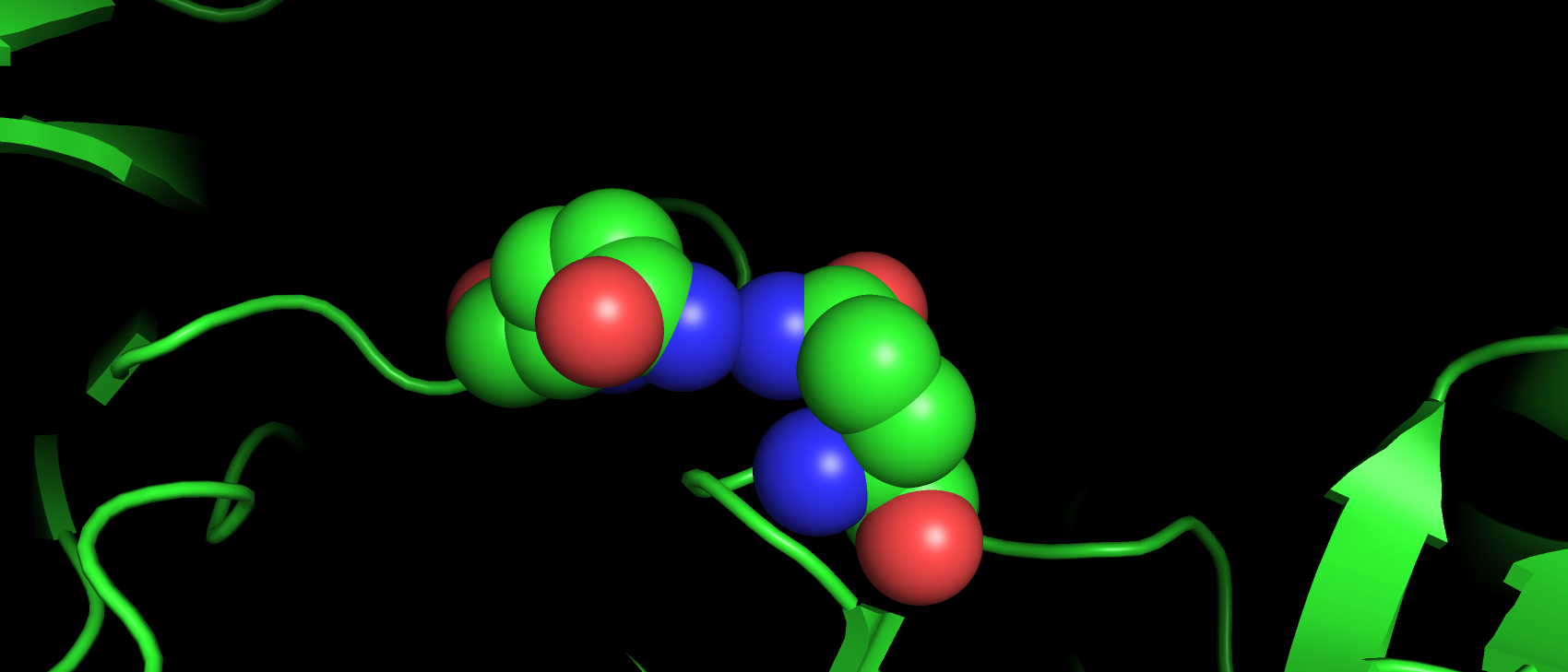
Дабы разнообразить примеры маргинальности (в структуре самого фермента маргиналов в принципе не очень много, как и
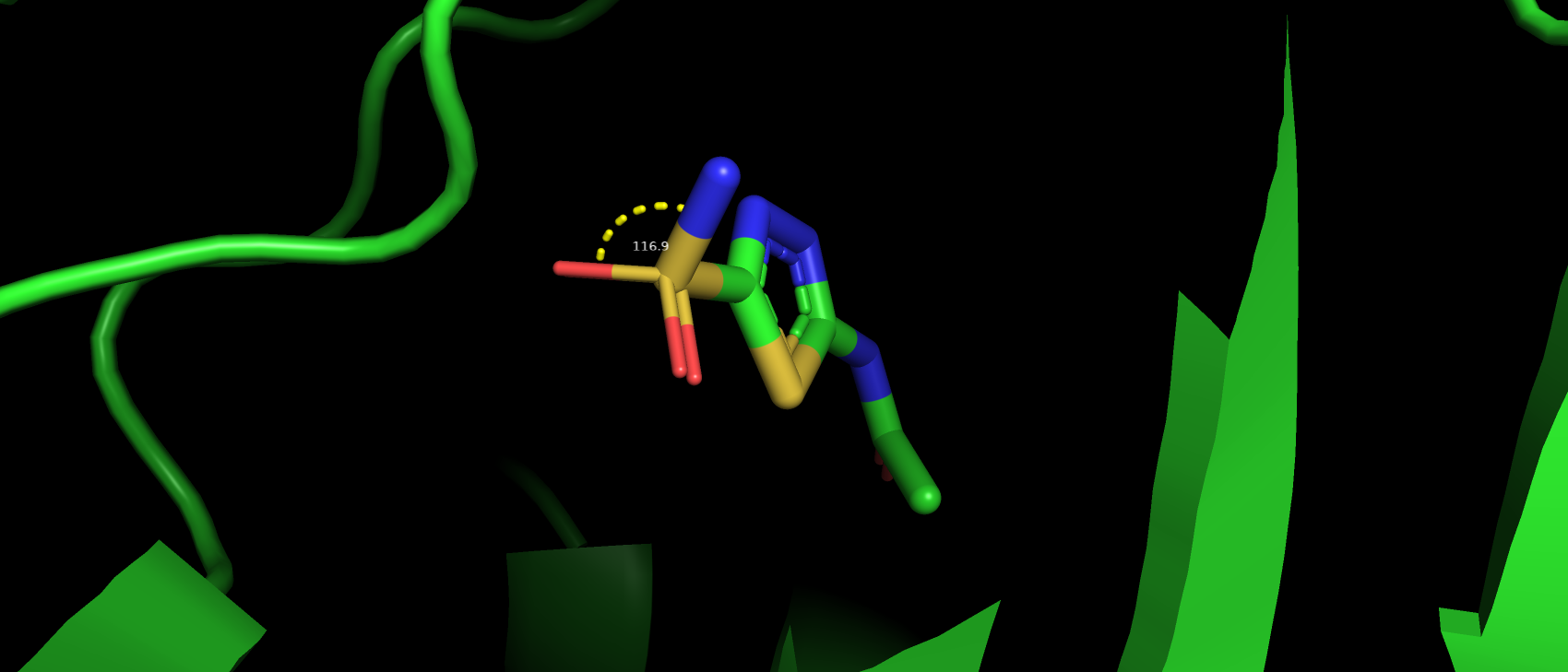
разновидностей критериев, по которым они найдены) я решил взять сам лиганд, так как в нем в отличии от структуры самого
белка наблюдаются аутлайеры по критерию угла ковалентной связи. Угол O2-S1-N1 имеет наблюдаемое значение 116.86, а
"идеальным" для него является значение 107.36.
Рис 10. Атомы, угол между которыми превышает "идеальные" значения в молекуле лиганда-ацетазоламида.
При запуске MolProbity при выполнении функции добавления водородов, выдается таблица позиций His/Asn/Gln
требующих разворота. Эта таблица полностью совпадает с таковой в репорте валидации PDB (13 маргинальных а.о.).
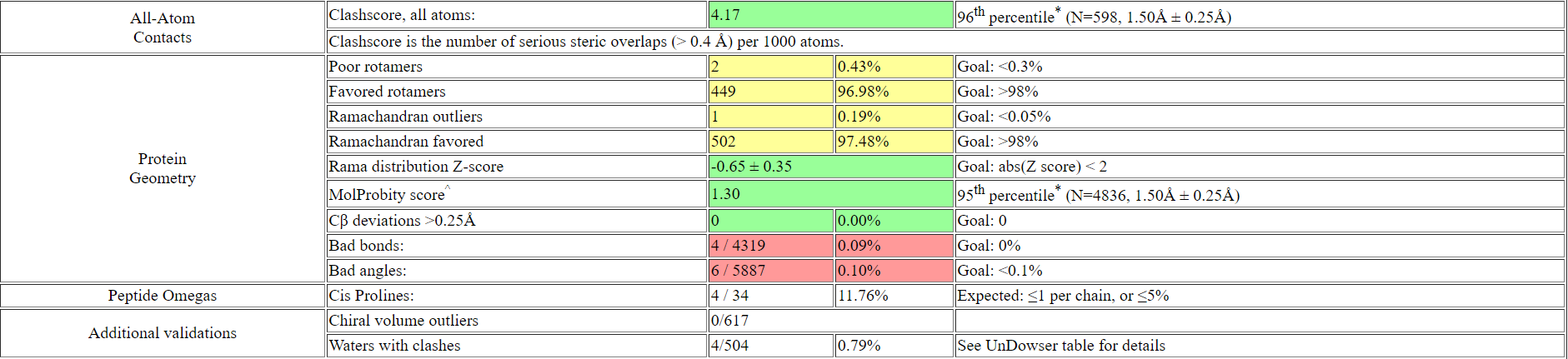
После добавления водородов и инверсии предложенных позиций количество клэшей на 1000 а.о. снизилось до 4.17.
Выдача MolProbity очень схожа с репортом валидации PDB. Остались одинаковыми количество аутлайеров по углам Ψ и Φ -
все также одинокий а.о. В:252:ASP, по углу chi сохранились 2 а.о. A:158:ASP и B:158:ASP. При этом заметны отличия:
MolProbity указал два аутлайера по длинам ковалентных связей в лиганде, которых нет в репорте валидации PDB; MolProbity
выдал меньше аутлайеров по углам ковалентных связей - всего 2, в то время как репорт валидации PDB содержит 11 (все они при
этом также находятся только у лиганда).
Рис 11. Выдача MolProbity после добавления водородов и инверсии предложенных а.о..
В моей структуре присутствует также ион цинка, поэтому дополнительно была проверена выдача CheckMyMetal.
Его выдача указывает, что два иона цинка имеют приемлимые значения критериев, связанных с геометрией, заселенностью и т.д.
Задание 3
После анализа выдач различных сервисов можно сделать вывод, что структура в целом хорошо подходит для изучения
функций фермента и его взаимодействие с лигандом-ингибитором. Это связано с тем, что структура имеет хорошее разрешение
с хорошим покрытием. Как было указано выше функциональные а.о. имеют хорошее ЭП и не являются маргиналами по каким-либо критериям.
Да и в целом количество маргиналов довольно маленькое. Так же важным в моем ферменте является хорошие показатели критериев
для ионов цинка.
Задание 4
Структуру из PDB-REDO я использовал, чтобы посмотреть как выглядят выбранные мной маргинальные остатки в переделанной модели,
есть ли какие-то улучшения в лучшую сторону.
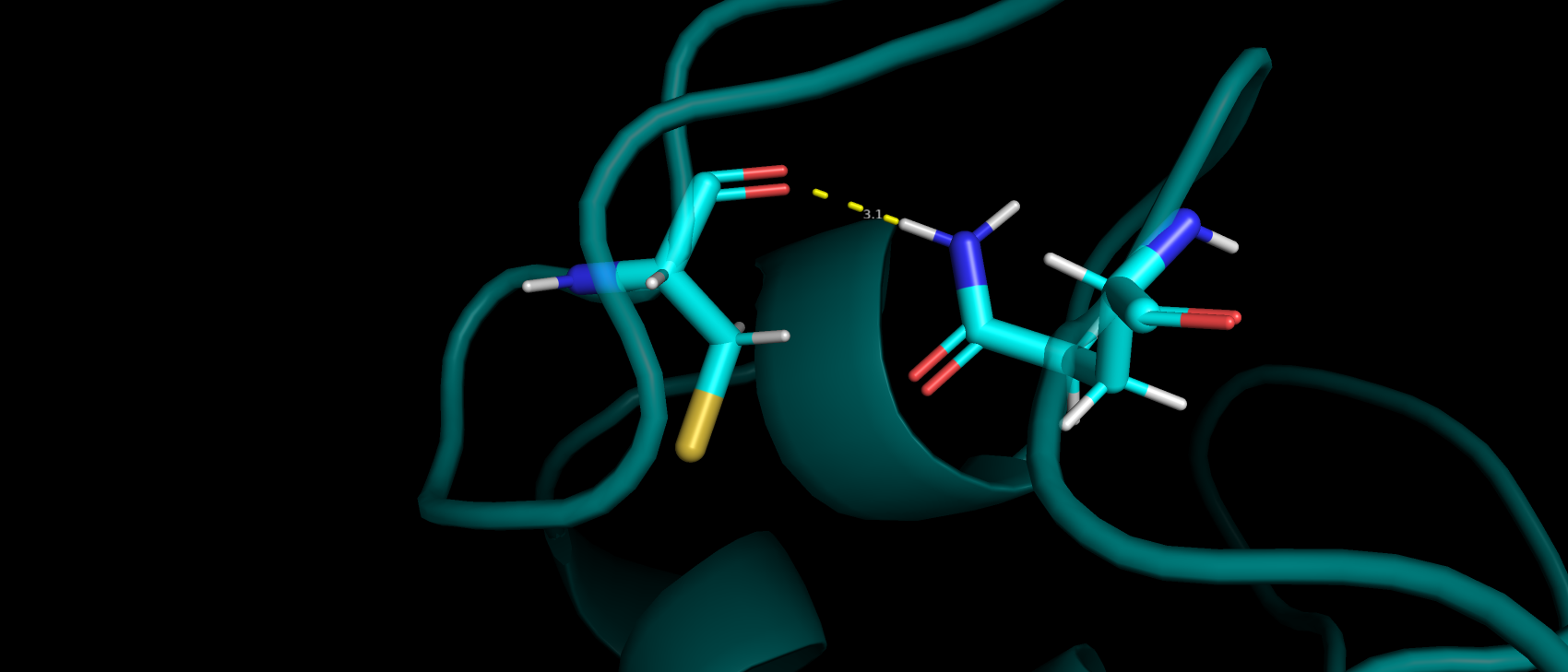
Первые маргиналы из задания 2 были A:28:GLN и A:203:CYS, у которых перекрывались Ван-дер-Вальсовы радиусы атомов водорода.
В структуре PDB-REDO в A:28:GLN был произведен разворот, в следствии чего в отличии от изначальной структуры в переделанной
между этими двумя остатками можно наблюдать водородную связь (рис. 12).
Рис 12. Водородная связь, появившаяся в переделанной структуре после разворота а.о. A:28:GLN.
Однако, для этих двух а.о. все равно наблюдается перекрывание Ван-дер-Вальсовых радиусов (рис. 13), хоть оно и куда
меньше, чем в первоначальной структуре.
Рис 13. Перекрывание Ван-дер-Вальсовых радиусов атомов A:28:GLN и A:203:CYS в переделанной структуре.
Однако появление водородной связи навязывалось в этом месте и при просмотре маргиналов в первоначальной структуре. Мне
кажется, что такое положение 2 аминокислот действительно лучше.
Следующим маргиналом был A:4:LYS, который имеет очень большой показатель RSRZ. Выдача PDB-REDO указывает, что для
этой аминокислоты было значительно лучше подогнана ЭП (это видно на рис. 14, где в отличии от
первоначальной структуры на уровне подрезки 1 появляется ЭП около атома азота в боковой цепи). Более того в структуре PDB-REDO
эта аминокислота уже не просто смотрит в раствор, а по-видимому образует водородную связь с A:ASP:11 (расстояние хорошее,
но угол немного смущает). Таким образом здесь, я тоже согласен с таким изменением.
Рис 14. ЭП вокруг A:4:LYS из переделанной структуры на уровне подрезки 1 и новое взаимодействие
с A:ASP:11.
Следующие маргиналы, где наблюдается пересечение Ван-дер-Вальсовых радиусов атомов азота A:112:GLN:NE2 и B:112:GLN:NE2.
В данном случае структура из PDB-REDO не отличается от первоначальной, а пересечение все также сохраняется. В целом это можно
объяснить тем, что от разворота как в первом примере ничего бы не изменилось, т.к. водородная связь здесь не образовывалась
бы из-за неудачного угла N-H-O, а азот и кислород по Ван-дер-Вальсовым радиусам очень схожи (рис. 15). По ЭП различить эти два атома
тоже довольно проблематично (рис. 16).
Рис 15. Сохранение пересечения Ван-дер-Вальсовых радиусов атомов азота A:112:GLN:NE2 и B:112:GLN:NE2
в переделанной структуре.
Рис 16. ЭП вокруг A:112:GLN и B:112:GLN из переделанной структуры на уровне подрезки 2.
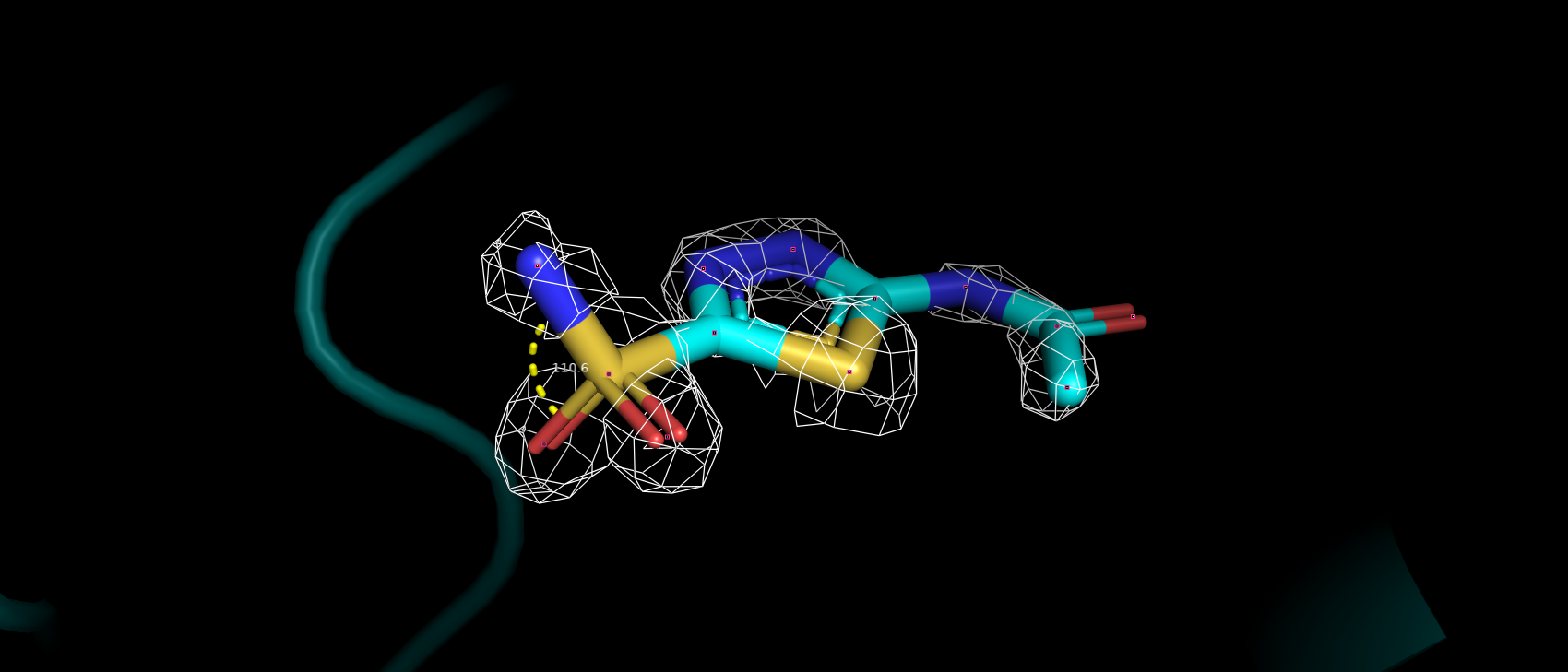
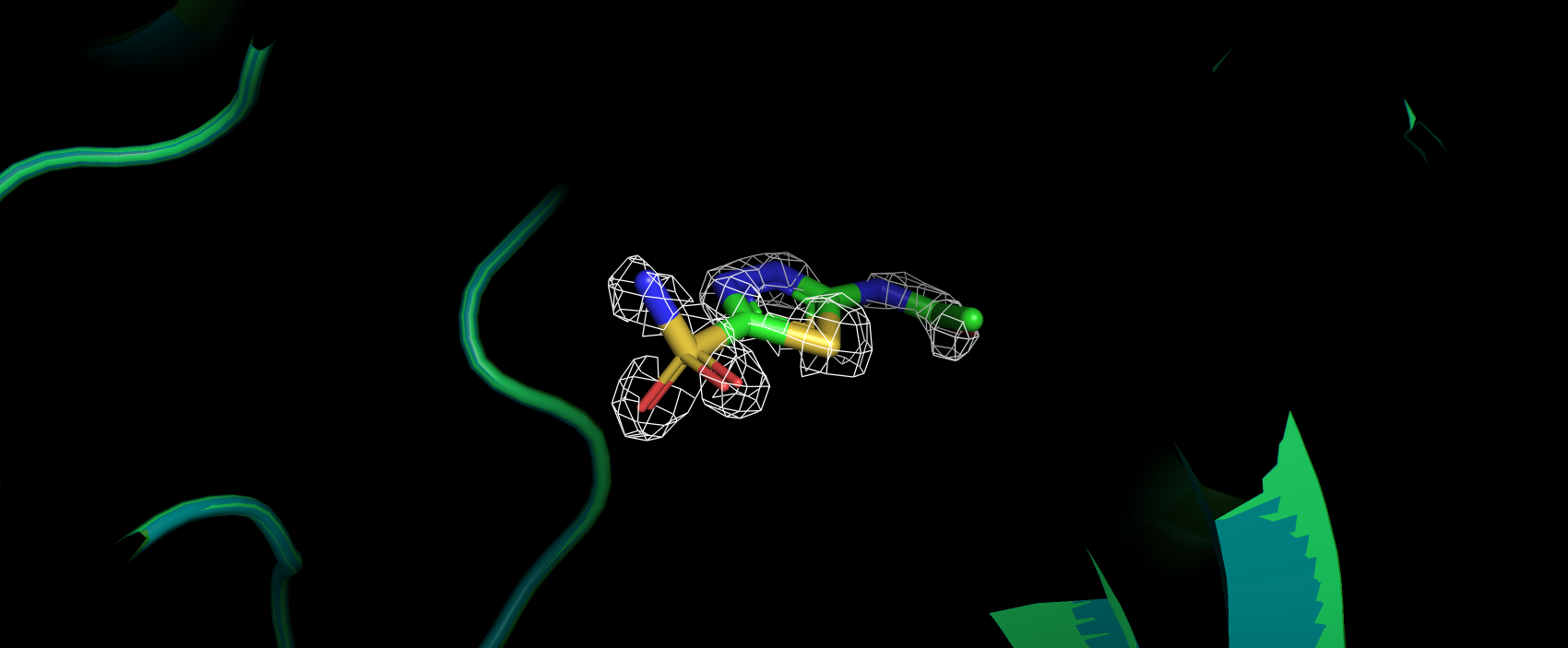
Также был представлен выше маргинал по углу ковалентных связей в ингибиторе. Угол O2-S1-N1 имеет наблюдаемое значение
116.86, а "идеальным" для него является значение 107.36. В структуре из PDB-REDO данный угол имеет значение 110.6,
что уже ближе к идеальному значению (рис. 17). Возможно, такая ошибка связана с недостаточно хорошим подгоном структуры лиганда в ЭП
в первоначальной структуре (рис. 18), на рисунке видно, как атом углерода выбивается из ЭП.
Рис 17. ЭП вокруг ингибитора в переделанной структуры на уровне подрезки 2.
Рис 18. ЭП вокруг ингибитора в первоначальной структуре на уровне подрезки 2.
Если же посмотреть по общим показателям, то структура PDB-REDO имеет меньшие значения R и R-free, 179 остатков были лучше
подогнаны под ЭП, 0 остатков изменились по ЭП в худшую сторону. 15 ротамеров было изменено. Так же можно отметить, что
структура в PDB-REDO имеет на 6 меньше удовлетворенных водородных связей. Как уже было сказано в задании 3 уже самого pdb
достаточно для изучения фермента. Однако структура из PDB-REDO тоже могла подойти для такой задачи. Также стоит отметить,
что раз в PDB-REDO ингибитор имеет более подогнанное под ЭП положение, возможно все-таки такая структура была бы лучше.