- вся структура
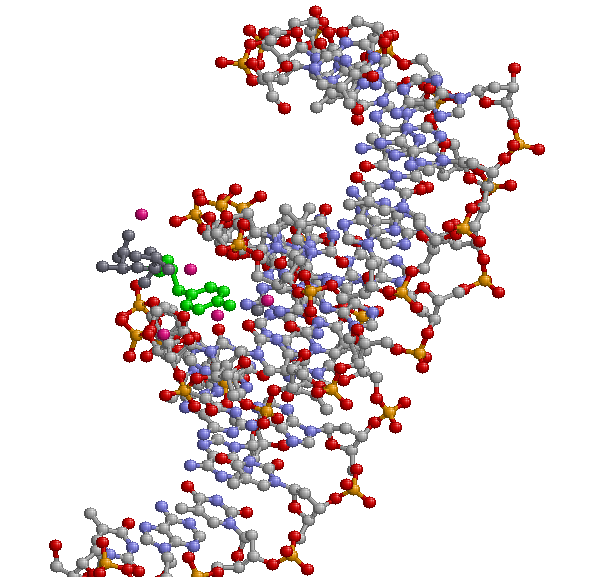
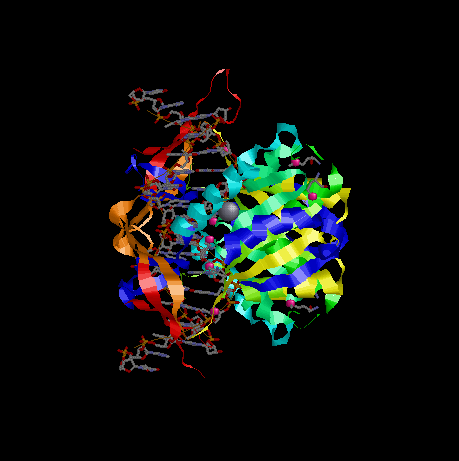 Рис.1
Рис.1 - только ДНК в проволочной модели
 Рис.2
Рис.2 - множество атомов set1
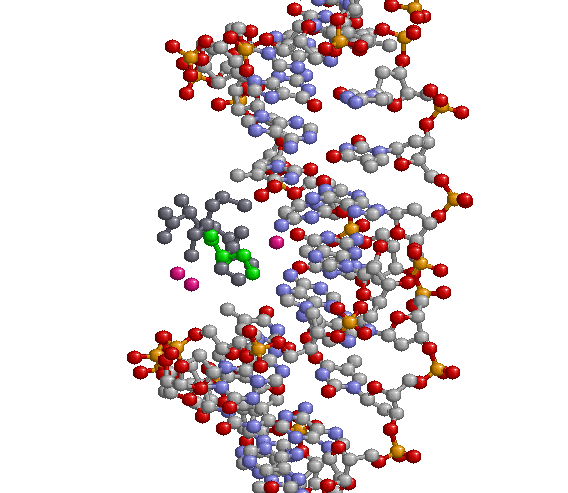
 Рис.3
Рис.3 - множество атомов set2
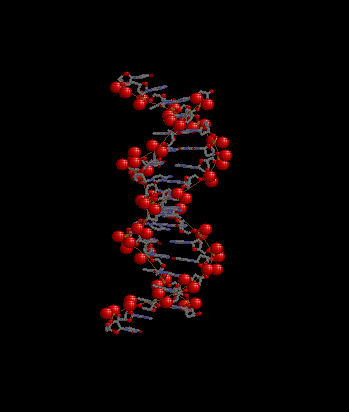 Рис.3
Рис.3 - множеством атомов set3
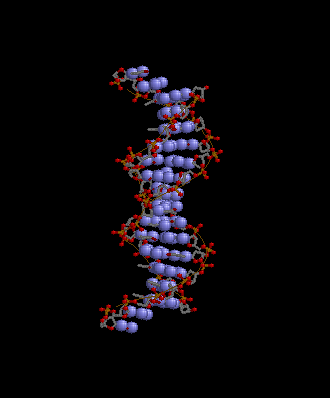 Рис.3
Рис.3 define o_in_phosphate *.O?P
|
Рис.1 |
|
Рис.2 |
|
Рис.3 |
|
Рис.3 |
|
Рис.3 |
| Контакты атомов белка с | Полярные | Неполярные | Всего |
| остатками 2'-дезоксирибозы | 7 | 65 | 72 |
| остатками фосфорной кислоты | 7 | 30 | 37 |
| остатками азотистых оснований со стороны большой бороздки | 0 | 2 | 2 |
| остатками азотистых оснований со стороны малой бороздки | 5 | 8 | 13 |
nucplot 1DFM_old.pdb
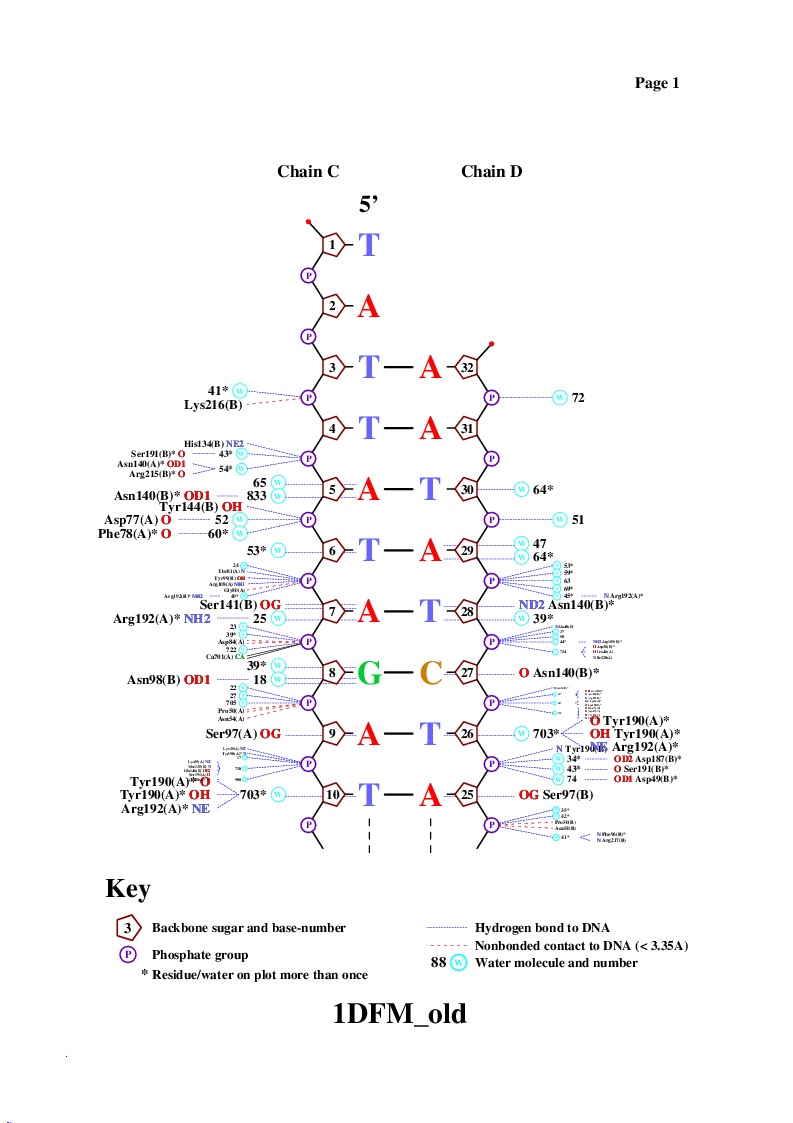
|
|
|
|
>2CV0:C|PDBID|CHAIN|SEQUENCE GGCCCCAUCGUCUAGCGGUUAGGACGCGGCCCUCUCAAGGCCGAAACGGGGGUUCGAUUCCCCCUGGGGUCACCA
einverted 2CV0.fasta
SEQUENCE: Score 15: 5/5 (100%) matches, 0 gaps
3 cccca 7
|||||
69 ggggt 65
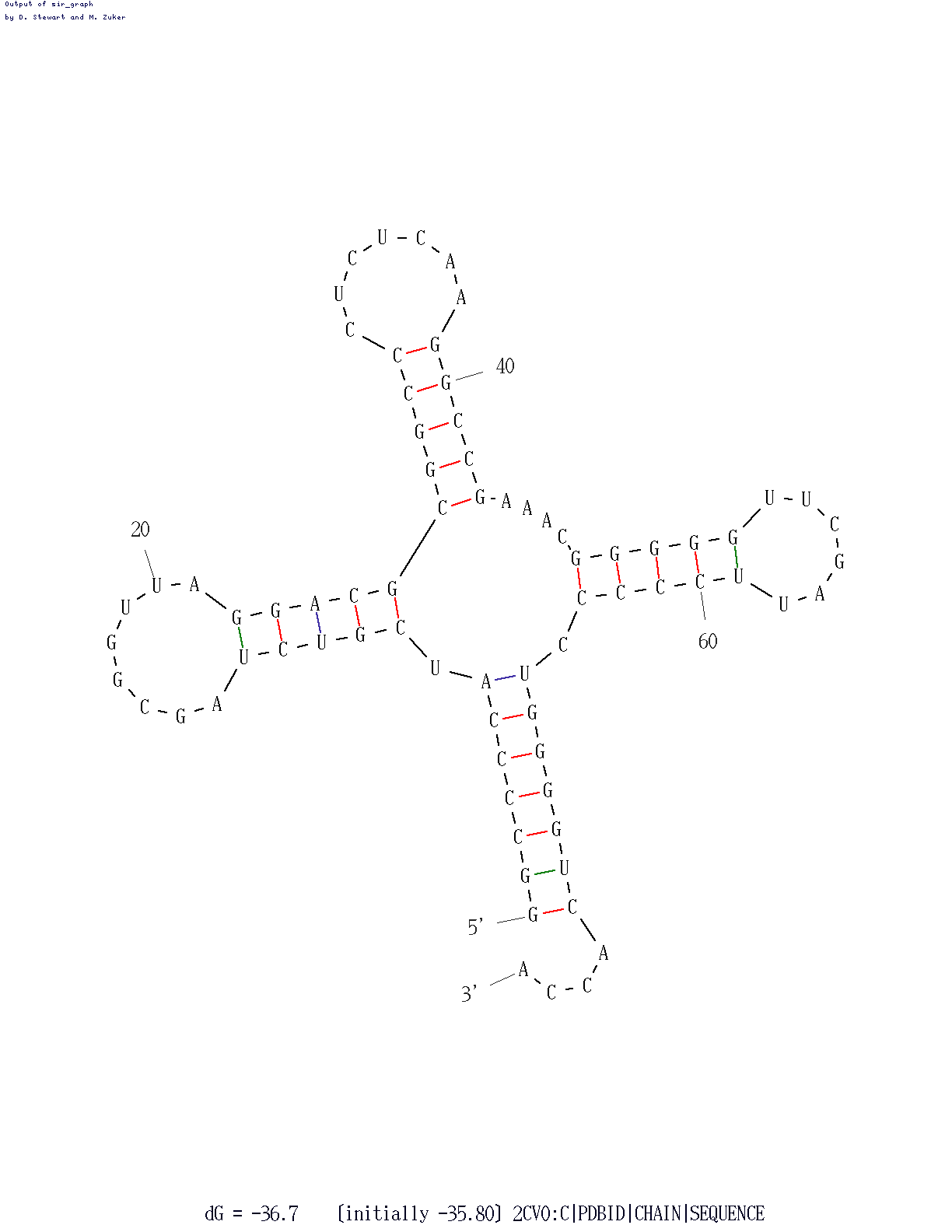
| Участок структуры | Позиции в структуре (по результатам find_pair) | Результаты предсказания с помощью einverted |
Результаты предсказания по алгоритму Зукера |
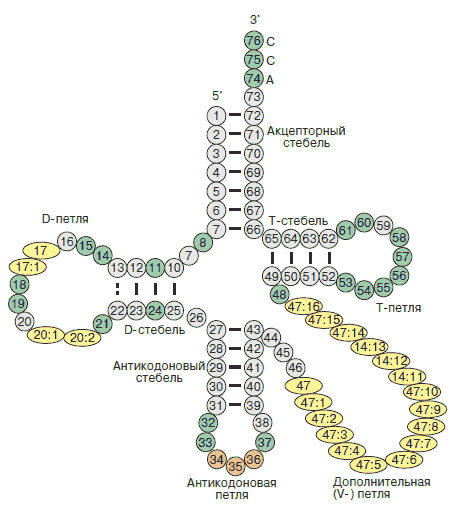
| Акцепторный стебель |
5'-501-507-3' 5'-566-572-3' Всего 7 пар |
предсказано 5 пар из 7 реальных | 7 из 7 |
| D-стебель |
5'-510-513-3' 5'-522-525-3' Всего 4 пары |
предсказано 0 пар из 4 реальных | 5 из 4 |
| T-стебель |
5'-549-553-3' 5'-561-565-3' Всего 5 пар |
предсказано 0 пар из 5 реальных | 5 из 5 |
| Антикодоновый стебель |
5'-526-532-3' 5'-538-544-3' Всего 7 пар |
предсказано 0 пар из 7 реальных | 7 из 7 |
| Общее число канонических пар нуклеотидов | 21 | 5 | 22 |