Из прошлого задания возьмем одно дерево, построенное методом neighbor-joining, например, с использованием PID.
Этот метод строит неукорененные деревья, в отличии от метода UPGMA.
Воспользуемся программой retree пакета PHYLIP. Для этого:
копируем файл retree.exe с диска P
копируем файл с деревом в Newick-формате в файл с именем intree (без расширения)
запускаем retree
выбираем укоренение в среднюю точку: "Midpoint root the tree" (M)
сохраняем получившееся дерево в Newick-формате в файле outtree
Получившеся дерево открываем программой MEGA.
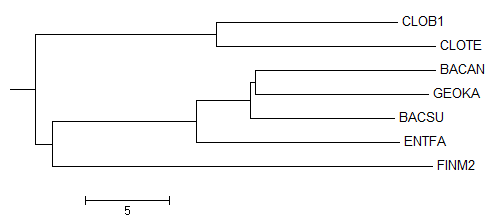
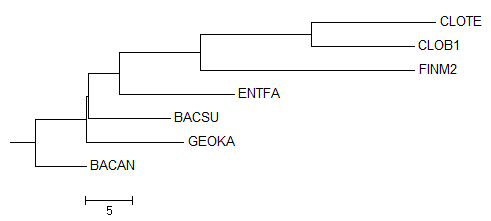
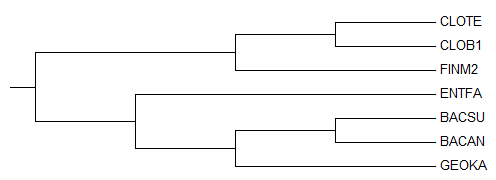
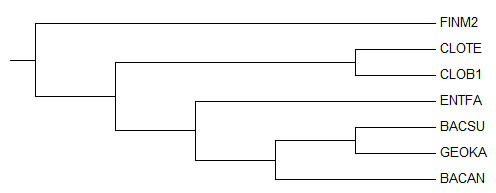
Дерево, укорененное PHYLYP в среднюю точку
Неукорененное дерево, до обработки PHYLYP
Исходное дерево, принятое за образец
По трем картинкам можно заметить, что изначально метод "neighbor-joining" привел неверное филогенетичеткое дерево
(отсутствует ветвь (BACSU,BACAN vs CLOTE,CLOB1,FINM2,ENTFA,GEOKA), вместо нее есть ветвь (BACAN,GEOKA vs CLOTE,CLOB1,FINM2,ENTFA,BACSU)).
Тем не менее остальные ветви проведены правильно, поэтому можно было ожидать, что укоренение произойдет в ветвь
(CLOTE,CLOB1,FINM2 vs BACSU,BACAN,GEOKA,ENTFA).
Вместо этого корень попал в ветвь (CLOTE,CLOB1 vs FINM2,BACSU,BACAN,GEOKA,ENTFA),
пусть и на совсем небольшом расстоянии от правильного попадания.
Можно предположить, что реальный корень находится вблизи FINM2, так как исходное дерево-образец построено без учета "молекулярных часов".
Задание 2. Укоренение с помощью внешней группы
Построим выравнивание методом максимальной экономии ("Maximum parsimony")
в MEGA. В данном случае невозможно укоренение в среднюю точку, так как метод не реконструирует длины ветвей.
Укоренение будем проводить с помощью белка E.Coli (по заданию).
Для этого выравняем соответствующие белки в Jalview, сохраним результат в FASTA-формате,
данные откроем MEGA (при импорте выбрать "Analyze").
Реконструируем дерево методом "Maximum Parsimony" (кнопка Phylogeny). Переукоренение в меню (Subtree / Root)
в ветвь, ведущую к E.Coli.
Чтобы получить изображение укоренённого дерева без ECOLI, нужно воспользоваться
кнопкой "Show Subtree Separately" (изображение голубой лупы на фоне дерева на левой панели окна MEGA).
Как мы можем видеть, данное дерево отличается от образца укоренением (в ветвь (FINM2 vs CLOTE,CLOB1,BACSU,BACAN,GEOKA,ENTFA) вместо (FINM2,CLOTE,CLOB1 vs BACSU,BACAN,GEOKA,ENTFA).
Кроме того, вместо ветви (BACSU,BACAN vs CLOTE,CLOB1,FINM2,ENTFA,GEOKA) здесь дается ветвь (BACSU,GEOKA vs CLOTE,CLOB1,FINM2,ENTFA,BACAN),
что не верно, однако, не должно сильно влиять на укоренение.
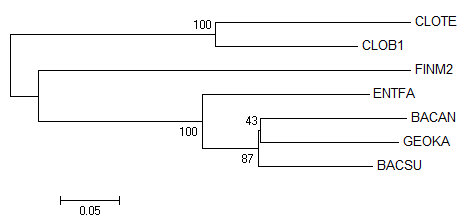
Задание 3. Бутстрэп-анализ.
Чтобы провести бутстрэп-анализ филогении белков, используя, например neighbor-joining метод, запускаем его из меню
программы MEGA.
Во всплывающем окошке которое открывается после этого,
в меню "Test of Phylogeny" выбераем
"Bootstrap method", число реплик, равное 100.
Полученное дерево не отличается от полученного ранее тем же методом без использования
бутстрэпа нисколько. Единственное, что появилось теперь - бутстрэп-поддержка (процент бутстрэп-копий
с данной ветвью).
"Original tree" и "Bootstrap consensus tree" отличаются тем, что "Bootstrap consensus tree"
не обладает длиной ветвей. Поддержка совпадает. В связи с этим, ограничимся изображением только одного из них.
По числам видно, что все правильные ветви имеют высокую поддержку, а ветвь, отделяющая
BACSU от BACAN, (BACSU,BACAN vs CLOTE,CLOB1,FINM2,ENTFA,GEOKA) достоверна лишь на 43%.
Так что у нас хотя бы появилась численная характеристика, позволяющая отделять достоверные ветви от
менее достоверных.